
FDA 和EMA 基于风险启动药品检查情形概述及对我国的启示
2024-01-08 08:56:18蔺娟
中国食品药品监管 2023年11期
蔺娟
国家药品监督管理局药品审评中心
乔利涛
国家药品监督管理局药品审评中心
周刚*
国家药品监督管理局药品审评中心
我国实施药品审评审批制度改革以来,国家药品监督管理部门陆续发布了一系列审评审批制度改革的配套文件[1-9],提出了优化审评审批流程,加快药品审评审批;建立以审评为中心,核查、检验为支撑的技术审评体系,逐步与国际接轨的药品监管科学体系等要求。提高药品审评审批效率,按时限审评成为常态要求,其中审评流程由省级食品药品监督管理部门受理、原国家食品药品监督管理总局审评审批的药品注册申请,调整为国家药品监督管理局集中受理。集中受理后,新受理的注册申请,将根据药品技术审评的需求由国家药品监督管理局食品药品审核查验中心统一组织全国药品注册检查资源实施现场核查[10]。注册核查模式由“逢审必查”向基于风险启动转变,基于风险启动药品注册核查是进一步优化注册程序的主要节点,是实现审评审批总时限可预期的重要保证[11]。
基于风险启动药品注册核查是适应药品高质量发展的需要,其既能够控制注册审评阶段对安全性、有效性、质量可控性的风险,又能够节约人力、物力,提高审评审批效率,提升行业的发展速度。因此,为了建立规范有效的合规审查体系,基于风险启动注册核查的原则和程序就显得尤为重要。我国已初步建立了基于风险启动注册核查的标准与程序,但仍在不断完善和提高,以期与国际接轨,建立更加科学、有效的适合我国国情的注册核查启动模式。本文介绍了美国食品药品监督管理局(Food and Drug Administration,FDA)以及欧洲药品管理局(European Medicines Agency,EMA)基于风 险启动批准前检查的情形和风险评估考虑因素,并提出了相关建议,为完善我国注册申请、启动注册核查提供参考与思路。
1 FDA 基于风险启动注册核查的概述
2002 年8 月21 日,FDA 启动了“21 世纪制药cGMP:基于风险的方法(Pharmaceutical cGMP for the 21st Century: A Risk-Based Approach)”倡议,提出鼓励实施基于风险的方法,将行业和机构的注意力集中到关键领域,确保监管审查和检查政策以最先进的制药科学为基 础[12]。2022 年9 月FDA 更新了《药品生产检查合规项目手册》(Compliance Program Guidance Mannual)中的《批准前检查手册(7346.832)》(7346.832Pre-approval Inspections-Investigations Revised)[13]旨在指导对某项申报是否需要进行现场检查建立标准,并制定了基于风险的决策标准,来判断是否需要对某项申请进行现场检查,从而保证检查资源使用到最有可能危害公众健康的领域。
1.1 基于风险的情形选择原则
2010 年版的《批准前检查手册(7346.832)》表明,FDA 的现场检查包括批准前检查、批准后监督检查、常规监督检查和有因检查。批准前检查(preapproval Inspection,PAI)指FDA 在药品申请审评过程中、药品批准之前,对药品申报中所提交的数据的真实性和准确性的核实,及对申请中列明的任何生产设施的现行药品生产管理规范(current Good Manufacture Practices,cGMP)状况通过现场检查和(或)地区文件审查的方式对生产企业进行的评估活动。进行PAI 有助于FDA 保证药品申请中所列生产企业具有生产药品的能力,及所提交数据的准确、完整。PAI 项目描述了3 种基本的检查目标,这些目标都要求综合信息策略以及仔细的现场检查评估:①为商业化生产的准备就绪;②与申报资料一致;③数据可靠性审计。
药品批准前检查的执行可分为2 种类型:优先检查和选择性检查。如果一个生产企业适用于一个或多个以下情况,则这个企业符合优先检查标准:某生产企业第一次被列入FDA 的注册申请中,即从未被检查过的企业或者仅针对非注册申请药物被检查过;申请人首次提交的注册申请(涵盖制剂生产和检验);已批准药品的首次仿制药申请(abbreviated new drug application,ANDA,涵盖制剂生产和检验);含有一种新分子实体的制剂(不适用于补充申请);制剂的含量测验范围较窄(如标识量95%~105% 的窄治疗指数的药物)或者需采取滴定法配药的药品(不适用于补充申请);制剂或原料药(active pharmaceutical ingredients,API)的生产工艺发生显著的变化或者采取了此生产企业认证之外的新剂型;API来源具有高风险(如API 来源于动物组织)或者预期用途发生了显著变化(如API 之前用于非无菌制剂,但现计划用于无菌制剂产品);提交了很多个申请,或者某些生产地点、工艺、产品的变更可能会对目前的设施或工艺的控制状态造成重大挑战;制剂或API 的申请状态分类为“不可接受”或者是因2 年内未经过现场检查,状态未更新(若为控制实验室此期限为3 年,若为包材与标签生产商此期限为4 年),或者是为初始申请,或者是化学、生产和控制(chemical manufacturing control,CMC)有重大变更的补充申请的批准前检查。
选择性检查则是除指定的检查任务外,其他不属于优先检查标准的情形。FDA 的生产和产品质量处(Division of Manufacturing and Product Quality,DMPQ)会在企业评估系统(establishment evaluation system,EES)中输入一个待评估的“10 日信”请求,并转发至地区办公室,根据地区办公室的建议确定是否进行批准前检查。对于FDA 地区办公室认为不需进行优先检查的,则将在10 日内把不启动批准前检查的原因与建议一起输入至EES 系统。对于建议需启动批准前检查的,地区办公室会使用EES 系统自动生成检查安排。
1.2 基于风险模型的风险评估
随着基于风险的方法不断成熟与完善,现行的《药品生产检查合规项目手册》强化了FDA 基于风险的方法,即利用申请中提供的信息和FDA 已有的关于该设施的信息确定是否需要进行检查。同时在原有PAI 项目描述的3 种基本检查目标的基础上增加了第4 个目标,即药品研发质量承诺,其意思是通过评估支持、定义、管理和持续评估其有效性以及在支持药品质量体系(pharmaceutical quality system,PQS)持续改进中的用途来评价药物开发计划。如果FDA 发现有足够的可用信息,则可能不需要PAI。当提交上市申请时,FDA 药品审评和研究中心(Center for Drug Evaluation and Research,CDER)通过组建整体质量评 估(integrated quality assessment,IQA)团队来执行质量评估,启动批准前设施评价。IQA 团队提供与药品相关的以患者为中心和基于风险的质量建议,包括对生产、加工、包装、贮存及检验药品或原料药的设施的建议。在执行质量评估时,IQA 团队通过评估以下内容来确定申请中列出的设施的PAI 需求:产品风险和生产工艺与设施风险以及申请中提供信息的准确性和可靠性。
产品知识和风险评估侧重于评估与具体产品使用背景(如治疗指数、患者人群、临床获益)下的产品关键质量属性(critical quality attributes,CQA)相关的风险。
生产工艺风险评估侧重于评估工艺对产品CQA 的影响,即关键变更参数源是否得到识别和解释,各维度的变更是否通过工艺得到了管理,以及工艺性能和产品质量属性是否可以充分满足并且有效控制。良好的产品和工艺评价意味着从患者的角度对质量至关重要的特征已被识别并转化为产品的CQA,并且影响CQA 的物料属性和工艺参数也能够识别、表征和控制。
生产设施风险评估侧重于评估生产或设施设备经证明的能力及其与上市申请的相关性,其包括但不限于通过评估设施检查报告(establishment inspection report,EIR)和依据,适用的现场警示报 告(field alert report,FAR),相 关的召回,监管、告诫行动以及可用的国外监管报告,审查设施近期的生产历史。
除以上3 个主要风险评估考虑外,对支持申请的生产场地信息的准确性和可靠性的评估,也是确定是否需要PAI 的一个重要因素。
另外,当需要确认质量数据(其对于确定药品的安全性、有效性和质量至关重要)的准确性和可靠性时或经评估需确认设施的操作与注册申请中申报操作是否相符也可触发PAI,最终也由IQA 团队根据对申请的综合风险评估来确定是否需要PAI。
1.3 注册核查启动流程
CDER 在收到新药申请(new drug application,NDA)或仿制 药申请(ANDA)的60 日内,药品生产评估办公室(Office of Pharmaceeufacturiong Assesment,OPMA)向监管事务办公室(Office of Regulatory Affairs,ORA)发送PAI 或地区文件审查(division of filing review,DFR)请 求,或通过CDER 的信息学平台软件Panorama 输入设施建议。对于PAI 请求:①OPMA通过OPMA 决策或请求任务以明确的理由要求PAI,并提供有关已确定的风险和疑虑的检查策略的特定信息。②ORA 评估请求,安排检查并通知OPMA。ORA和CDER 应尽可能在应用评估和检查活动的计划和时间安排上进行合作。如果ORA 的评估表明没有必要进行PAI,则需与OPMA 共同做出最终决定。在收到请求的10 个工作日内,ORA 的批准前项目经理(preapproval program manager,PAM)需 在Panorama 中 录入不启动检查的原因以及ORA 的建议。
2 EMA 基于风险启动注册核查的概述
欧盟是由多个成员国组成,药品的审批程序既要考虑经济一体化的整体性,又要兼顾各成员国独立自主的个体性,因此其药品审批程序更多样化,主要分为成员国审批程序、集中审批程序、互认可程序。2001 年,欧盟对这些指令进行了梳理整合,最终由欧洲议会和欧盟理事会通过了2001/83/EC 号法令《关于人类用药品的共同体法规》[14],该法令是目前欧盟最全面、最系统的药事法规。2013 年修订了附件1 第四部分,引入了“基于风险的方法”。
2.1 基于风险的情形选择原则
2022 年4 月20 日《欧盟检查和信息交换程序汇编》(Compilation of Community Procedures on Inspections and Exchange of Information)进 行了修订[15],其中《基于风险的药品生产企业检查计划模型》(A Model for Risk Based Planning for Inspections of Pharmaceutical Manufacturers)提出了通过使用系统的、基于风险的方法,并充分利用其监督和执法资源,用于在规划药品生产质量管理规范(Good Manufacture Practices,GMP)检查的频率和范围时确定检查地点的优先顺序。《基于风险的药品生产企业检查计划模型》适用范围为成员国主管当局对制剂和原料药生产企业的GMP 检查计划,国内和第三国(third country manufacturers)生产企业的GMP 检查计划,成员国主管当局对研究用药品生产企业的GMP 检查计划。但是对于新的生产场地,如果第三国没有签署双边互认协议(mutual recognition agreements,MRA)并且工厂从未接受过欧盟检查员检查,则将启动系统地检查。对于既往已接受过检查的,将根据不同生产场地的风险进行评级,其主要基于2 种类型风险的评估,即内在风险和合规风险。
内在风险,主要是指生产场地的复杂性(规模和设施种类)、产品制造过程的复杂性(操作类型和顺序过程控制)和产品复杂性,以及生产场地提供的产品或服务的关键性。值得注意的是,这些项目的复杂性和关键性通常保持不变。生产场地的复杂性主要包括:生产场地规模的复杂性;现场使用的不同制造或分销流程的数量;现有设备和设施的配置程度;现场工作人员的数量;企业生产提供的商业市场数量;企业生产提供的客户数量。产品制造过程复杂性主要包括:无菌和无菌制造工艺;有高度复杂过程的参数放行程序;具有大量关键步骤的生产工艺;品种剂型为低剂量高溶解性速释剂、缓释剂等制造过程的复杂制剂;非无菌制造过程中的单元操作数量较大导致过程复杂的;在生产现场进行的后处理或返工的程度。产品复杂性主要包括:包装组件数量较多的产品;需要特殊储存和配送的产品。
合规风险,是基于最近一次GMP 检查的结果,主要考虑因素为缺陷的分类和数量,反映了企业执行GMP 的合规水平,以及GMP 合规状态等级。合规风险的主要考虑因素包括:在最近一次现场检查中发现缺陷的区域,特别是严重和主要缺陷;最近一次现场检查中未检查(或未详细检查)的区域;上次检查时认为资源不足的区域;生产场地的计划更改可能会改变与生产场地相关的复杂性或关键性风险评级;检查员认为有必要在下一次检查的。
2.2 基于不同类型风险的评估
根据内在风险考虑的2 个风险指示因素,即生产场地、产品制造过程和产品的复杂性以及生产场地提供的产品或服务的关键性。通过每个品种内在风险的估计值的固有风险矩阵可最终判定品种的内在风险等级,见表1。
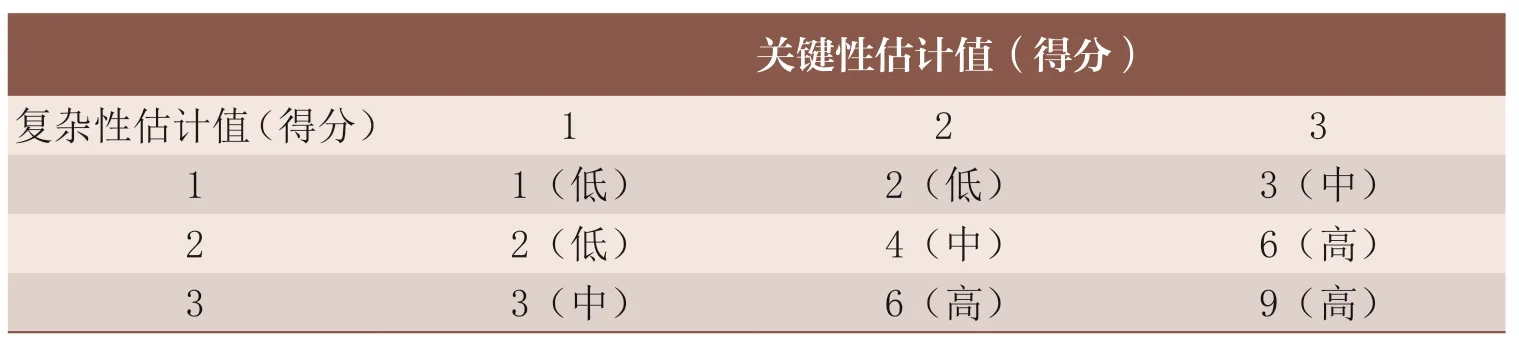
表1 每个品种内在风险的估计值的固有风险矩阵表
合规风险的评估是基于最近一次现场检查(例行或全面检查,有因检查)中与生产场地相关的合规风险,主要是通过现场检查中发现的缺陷,对其进行梳理分级,分配相应的高、中或低的等级,然后再对现场检查进行合规风险评级,见表2。

表2 现场检查合规风险评分表
将内在风险和合规风险利用简单的矩阵进行组合后生成最终的生产场地的检查风险等级,见表3。该结果可用于决定下一次现场检查的频次和范围。

表3 总体风险评矩阵表
2.3 基于风险的检查频率
根据生产场地的检查风险等级评级来生成和记录现场例行检查的建议频率,每个风险评级的建议检查频率,见表4。

表4 每个风险评级的建议检查频率
风险评级A 的生产场地在固有风险或合规风险方面至少有一个低风险评分。在例行检查计划中,这些场所的检查频率可能会降低,如频率低于每2 年一次。
风险评级为B 的生产场地,检查频率的时间范围设定为 1~2 年,但不是绝对的 2 年。例如,如果2 个生产场地的风险评级为 B,但其中一个的最后检查结果比另一个差(比如存在5 个主要缺陷与一个主要缺陷),则分配给前一个生产场地的检查频率原则上为检查频率规定的时间范围的下限(检查频率接近1 年而不是2 年)。
风险评级C 的生产场地在固有风险或合规风险方面至少有一个高风险评分。在例行检查计划中,可能会增加检查这些生产场地的频率,至少每年一次或更多的频率。
此外,分配给具有相同风险评级的生产场地的检查频率可能会考虑内在风险和合规风险各自的风险估计值的分数高低,从而最终确定该生产场地的检查频率。例如,对于总体风险评级都为C 的生产场地,当一个生产场地同时具有高内在风险和高合规风险时分配的检查频率(例如9 个月)可能高于具有高内在风险但中等合规风险的生产场地。
上述风险评级目的是保证具有高内在风险评分或高合规风险评分的生产场地都不会被分配降低的检查频率。分配给任何一个生产场地的实际检查频率都能反映上次检查期间发现的缺陷的数量和类型。
3 我国现场注册核查启动程序的现状
3.1 基于风险的情形选择原则
《药品注册管理办法》[16]第四十六条规定“药品审评中心根据药物创新程度、药物研究机构既往接受核查情况等,基于风险决定是否开展药品注册研制现场核查”,第四十七条规定“药品审评中心根据申报注册的品种、工艺、设施、既往接受核查情况等因素,基于风险决定是否启动药品注册生产现场核查”,其配套文件《药品注册核查检验启动工作程序(试行)》[17]基于品种因素和研发生产主体合规因素,对需纳入注册核查风险等级划分的情形进行了规定:①药品上市许可申请;②涉及药品生产过程中处方工艺或生产批量重大变更,或者新增临床试验数据等的补充申请;③其他需要启动注册核查的药品注册申请。
品种因素包括药物创新程度、药品类型、工艺和设施等。研发生产主体合规因素包括参与药学研制、临床试验、药理毒理学研究以及生产制造的相关单位和机构既往接受注册核查与监督检查的情况等。在此基础上,将品种和研发生产主体合规两大类评估因素分别划分为高、中、低3种情形。根据品种因素和研发生产主体合规风险因素具体情形,经综合评估研判后,药品审评机构将需要注册核查的药品注册申请划分为高、中、低3 个风险等级,原则上以品种因素和研发生产主体合规因素风险情形较高的确定药品注册申请最终风险等级。
3.2 注册核查启动流程
药品注册申请原则上在受理后40 日内,由药品审评机构完成品种因素以及合规因素的风险标记,经综合评估后确定品种注册核查风险等级及核查类别(药学、临床),其中对于高风险等级的药品注册申请应当启动注册核查,对于其他风险等级的药品注册申请按比例随机启动注册核查。对于确定启动注册核查的药品注册申请,由药品审评机构准备注册核查所需相关资料,并在规定时间内通知核查机构启动注册核查,同时通知申请人,完成启动注册核查任务。
4 对我国现行药品注册核查启动标准与程序的思考
目前,我国药品注册申请基于风险启动注册核查的风险因素考虑方面与发达国家和地区的监管机构启动注册核查的考虑因素类似,均为通过品种因素(药物创新程度、品种、工艺、设施等)和合规因素(研发生产主体既往接受检查情况)两方面的风险考虑因素,经综合评估后研判注册申请的最终核查风险等级。诚然,我国的药品注册核查启动工作程序已经在多个方面与国际先进水平接轨,但在进一步提升认知风险、识别风险的能力,分层次、有步骤地进行风险识别、分析和调整等方面仍需完善与提高。探索建立符合我国国情的基于风险启动核查的合规风险评估模型,不断完善符合我国国情的药品注册核查启动标准与程序,构建药品注册核查启动前基于风险评估的合规审查体系,才能充分适应不断发展、不断进步的药品监管理念,提高科学监管能力,保障公众用药安全。
猜你喜欢
江苏安全生产(2022年10期)2022-11-02 09:37:32
民用飞机设计与研究(2020年4期)2021-01-21 09:15:00
林业科技(2020年3期)2021-01-21 08:28:52
现代经济信息(2020年34期)2020-06-08 06:02:18
中国外汇(2019年20期)2019-11-25 09:54:54
中国外汇(2019年16期)2019-11-16 09:27:40
中国化肥信息(2018年3期)2018-01-30 06:56:43
知识经济·中国直销(2017年3期)2017-04-16 03:07:53
现代企业(2015年4期)2015-02-28 18:48:39
环球时报(2014-08-02)2014-08-02 08:26:42
