
使用中间层受监督的自编码器探索蛋白质的构象空间*
2024-01-06 10:24陈光临张志勇
物理学报 2023年24期
陈光临 张志勇
(中国科学技术大学物理系,合肥 230026)
1 引言
蛋白质的功能与其结构和动态构象变化密切相关[1].为了获得蛋白质分子的结构,人们开发出了各种实验和预测技术.X射线晶体衍射[2]和冷冻电镜技术[3]可以解析高分辨率的蛋白质分子结构,而核磁共振[4]可以提供分子中的原子距离等信息.此外,小角X射线散射[5]、化学交联[6]和荧光共振能量转移[7]等技术可以从不同的角度给出蛋白质分子的各种结构信息.基于人工智能的结构预测方法,如AlphaFold2[8]和RoseTTAFold[9],可以直接根据氨基酸序列预测蛋白质的结构.这些方法在获取蛋白质静态结构时十分有效,但是不易得到蛋白质的动态变化信息.
计算模拟方法,例如分子动力学(molecular dynamics,MD)模拟,是研究蛋白质分子动态变化的重要工具[10].MD方法用半经验的能量函数来描述原子间的相互作用,在经典力学的框架下对蛋白质分子进行模拟.从一个已知的分子结构出发,通过迭代求解运动方程,得到分子动态变化的过程.为了确保结果的可靠性,通常要求对整个构象空间充分采样.但由于分子模拟的结果服从玻尔兹曼统计,在生理条件下,对高能构象的采样十分困难,这一问题通常需要引入增强采样等方法来解决[11].模拟的另一个问题来自分子力场,它是对分子间相互作用的一种近似描述,因而必然存在一定的误差.力场选择不合适可能会导致模拟结果表现出与实际情况不同的倾向[12],即使经过大量计算后达到了充分采样的要求,也无法正确描述生物大分子的动态变化.这种情况下,可以先尽可能多地产生不同的构象,再验证其合理性.
近年来,机器学习方法的快速发展为解决分子模拟中的采样和力场问题提供了新思路[13,14].自编码器是一种生成神经网络,最初用于计算机图形领域[15],目前也应用于探索蛋白质分子的构象空间[16].自编码器由编码器和解码器组成,高维的蛋白质结构信息经过编码器压缩得到低维空间的隐变量,再经过解码器重构出蛋白质结构,同时要求重构的结构与输入的结构尽可能一致.训练完成后,只需要向解码器输入随机数据,就可以构建出不同的蛋白质构象.由于自编码器在训练过程中只要求数据成功重构,中间层的隐变量没有明确的含义,而构象生成是从中间层的数据开始的,因此探索构象空间的方向也是不确定的,有时可以找到各种不同的构象,有时只能得到不感兴趣或不合理的构象.为了解决上述问题,一种常用的方案是对中间层的结果进行一些限制.
本研究设计了一个有监督的自编码器模型.将一些反应坐标引入到自编码器中,要求其在重构蛋白质结构的同时,中间层的数据要与给定的反应坐标接近,从而使构象空间的探索在给定的方向上进行.将该模型运用到两个多结构域蛋白,噬菌体T4溶菌酶和腺苷酸激酶,探索得到的蛋白质构象空间覆盖了目前已知的实验结构.通过引入合理的反应坐标和实验数据,建立有监督的自编码器模型,有望成为探索蛋白质构象空间的有效工具.
2 方 法
2.1 中间层受监督的自编码器模型
为了实现在给定方向的构象空间探索,使用Pytorch2.0设计了一个中间层受监督的自编码器(图1).该模型的整体结构与普通的自编码器相似,由编码器和解码器组成.其中编码器是一个多层的全连接神经网络,在输入层之后每一层的维数分别是2048,512,128,32,8,2,解码器也是多层全连接网络,其结构与编码器对称,每一层的维数依次是2,8,32,128,512,2048,输出层的维数与编码器输入层相同.除了最后一层外,编码器和解码器的每一层都使用了ReLU作为激活函数,而最后一层则使用Sigmoid激活函数,以控制输出结果的范围.这一模型的参数量很少,对计算资源的要求较低.

图1 中间层受监督的自编码器示意图Fig.1.Schematic of supervised-AE.
不同于无监督的自编码器,将监督学习引入自编码器的中间层,训练时使用的损失函数形式如下:
其中Loutput为输出层的损失函数,用来描述重构后的结构与输入结构之间的差距;Lmidden为中间层的损失函数,描述中间层数据与输入结构对应的反应坐标之间的差距.只使用反应坐标往往不能准确地描述和重构整个分子结构,只能反映结构的某些特征,因此模型需要在正确提取反应坐标和成功重构分子结构之间找到平衡.引入了权重因子ω来调整两者对损失函数的贡献,ω较大时,中间层对损失函数的贡献更大,模型会倾向得到给定的反应坐标,而重构分子结构的效果会变差,反之,ω较小时,模型可以完成分子结构的重构,但中间层的数值不一定接近给定的反应坐标.本文中,该因子的值设定为100.
2.2 数据获取
训练模型的数据来自MD模拟.模拟的体系分别是噬菌体T4溶菌酶(T4 lysozyme,T4L)和大肠杆菌腺苷酸激酶(adenylate kinase,AdK).T4L及其突变体在PDB数据库中有大量晶体结构,其结构变化主要体现在N端结构域和C端结构域之间口袋的打开和关闭(图2(a)).AdK可以分为CORE,LID以及AMPbd三个结构域,分别在CORE和LID,以及CORE和AMPbd之间形成两个口袋.在酶的催化过程中,口袋的打开和关闭十分重要(图2(b)).这两个蛋白分子的动态构象变化已经研究得比较充分,适合用来验证我们的模型.

图2 本研究中使用的两种蛋白质分子的不同结构 (a) T4L的闭合(不透明)和打开(透明)结构,紫色为α螺旋,黄色为β折叠;(b) AdK的闭合(不透明)和打开(透明)结构,不同颜色表示不同的结构域Fig.2.Different structures of the two proteins in the work.(a) The close (opaque) and open (transparent) state of T4L.α-helix is colored in purple and β-sheet is colored in yellow.(b) The close (opaque) and open (transparent) state of AdK.Different domains are colored in different colors.
根据蛋白质分子的结构变化特征,计算相应的反应坐标作为监督引入到自编码器的中间层.从T4L及其突变体的晶体结构中选取能够反映其构象变化的41个结构,消除它们之间的平动和转动后,使用Cα原子的坐标进行主成分分析.特征值最大的2个主成分分别对应T4L的开闭和扭转运动,其占比分别为86%和6%.因此使用这2个主成分作为反应坐标,可以较好地描述T4L分子的运动[17].AdK的结构变化主要表现为结构域的相对运动,因此可以选取CORE-LID和COREAMPbd结构域的质心距离作为反应坐标[18].
分子动力学模拟使用GROMACS-2023版本进行[19].从PDB数据库中分别选取T4L的打开(PDB编号2LZM[20])和关闭(PDB编号178L[21])结构,以及AdK的打开(PDB编号1AKE[22])和关闭(PDB编号4AKE[23])结构作为模拟的初始构象.为了验证模型是否受分子力场的影响,每一组模拟都分别使用了AMBER99SB力场/OPC水模型的组合[24,25]以及CHARMM36m力场/TIP3P水模型的组合[26].将分子放入正十二面体的周期性盒子中,同一分子的不同体系使用同样大小的盒子,以避免盒子尺寸对模拟结果的影响.向体系中填充水分子,并加入离子直到电荷平衡.先后用2000步最速下降法和1000步共轭梯度法进行能量最小化,然后在NPT系综下进行100 ps的位置约束MD模拟,以平衡系统的温度和压强,随后进行NPT模拟以获取训练模型的数据.AdK在没有结合配体时无法维持关闭状态,因此在模拟中额外加入了结构域距离的位置限制.所有模拟的步长均为2 fs,使用LINCS算法约束氢原子参与的化学键,分别用V-rescale[27]和C-rescale算法控制系统的温度和压强,非键相互作用中静电相互作用通过PME[28]算法计算,范德瓦耳斯力则做截断处理,截断距离为1 nm.
由于不要求充分采样,每组用于产生训练数据的模拟仅进行100 ns,每10 ps保存一个结构,共保存10000个.消除不同结构之间的平动和转动变化后,提取主链部分的原子,即N,Cα,C,O的笛卡尔坐标作为模型的输入,同时计算出每个结构的反应坐标作为标签.在开始训练之前,还需要对数据进行归一化处理,数据的每一个维度都分别被放缩到0.2与0.8之间,这一区间Sigmoid函数的斜率较大,有利于模型训练更快达到收敛.
2.3 利用有监督的自编码器探索蛋白质构象空间
将模拟轨迹整理为数据集,从中随机取出80%作为训练集,剩余的20%作为测试集.以平方误差作为损失函数,用Adam优化器[29]进行训练,遍历训练集500次,初始学习率为 1×10-4,并随着遍历次数以 1×10-8的速率减小.完成训练后,在 [0.05,0.95]×[0.05,0.95] 的范围内均匀选取10000个点作为自编码器中间层隐空间的数据点,将这些点输入解码器构建出对应的蛋白质分子主链结构.模型训练和数据生成的相关运算在RTX 3090Ti上运行.
由于生成的结构并不总是合理的,通过两种判据对其进行筛选.其一是蛋白质的主链二面角取值需要满足一定的规律,这一规律通常用Ramachandran图来描述,将大量已知蛋白质结构的Ramachandran 图的统计结果[30]作为参考,与模型生成的蛋白质结构的Ramachandran 图进行比较,若90%以上处于合理区间,则认为该结构的主链二面角分布是合理的.其二是不同原子之间不能存在空间冲突,使用分子模拟工具OpenMM[31]对分子结构进行一小段能量最小化,如果最终原子间的力比较小,就可以认为该分子不存在空间冲突.考虑到这一步需要频繁进行,与其他分子模拟工具相比,使用直接运行在Python中的OpenMM可以节省大量用于初始化模拟引擎的时间.由于模型仅产生主链部分的原子坐标,并非完整的分子,用ParmEd工具[32]将力场参数中非主链的部分删去,同时将所有原子的电荷设置为0,在能量最小化时仅保留化学键和范德瓦耳斯力.能量最小化不仅可以筛选掉明显不合理的结构,还可以对结构中的一些键长键角的错误进行修正.
模拟得到的构象空间分布十分有限,在此基础上进行构象空间探索也因此受到限制.为了进一步扩大构象空间探索的范围,将模型生成的结构与原有数据集的一半合并成新的数据集,并重复进行模型训练和构象空间探索.随着探索范围逐渐扩大,模型生成的不合理结构逐渐增加,构象空间的探索效率也随之下降,因此只重复上述流程3次.
3 结果与讨论
3.1 T4L构象空间探索结果
以T4L的模拟轨迹作为训练集,进行训练以及构象空间探索,整个流程耗时仅20 min.探索结果如图3所示,由于使用不同力场得到的模拟轨迹不同,构象空间探索的区域也有所不同,整体上看使用AMBER99SB力场/OPC水模型的探索范围更大.不过使用两种力场得到的探索范围都可以覆盖包括所有参考晶体结构在内的训练集附近的区域,例如可以找到与PDB编号为173L晶体结构[21]十分相似的构象(图4(a)),RMSD为0.7 Å.此外,探索结果中还可以看到大幅度的构象变化,例如闭合状态与打开状态的不同(图4(b)),以及两个结构域的相对转动角度不同(图4(c)).

图3 T4L的构象空间探索结果 (a) 使用AMBER99SB力场/OPC水模型;(b)使用CHARMM36m力场/TIP3P水模型Fig.3.Results of conformational space exploration of T4L:(a) With AMBER99SB/OPC;(b) with CHARMM36m/TIP3P.

图4 探索到的不同T4L构象 (a) PDB编号173L的晶体结构(不透明)与探索到的相似结构(透明);(b) 开合程度不同的两个构象;(c) 扭动情况不同的两个构象;紫色为α螺旋,黄色为β折叠Fig.4.Different T4L conformations explored: (a) PDB:173L(opaque) and a similar structure explored;(b) two conformations with different degrees of opening and closing;(c) two conformations with different degrees of twisting.α-helix is colored in purple and β-sheet is colored in yellow.
虽然模型生成的结构都通过了二面角分布的检验,以及键长键角和空间冲突的修正,但依然存在一些不合理的情况,如生成的结构中二级结构含量显著低于晶体结构和模拟轨迹中二级结构的含量.为了验证模型产生结构的合理性,我们使用kmeans算法,根据反应坐标将探索结果分为50组,取每一组最靠近中心的构象作为代表,用tleap补全侧链,然后进行100 ns约束Cα原子的MD模拟,从而在不改变反应坐标的情况下修复二级结构.除少数情况由于侧链存在空间冲突而失败外,大部分代表构象的二级结构得到修复(图5(a)和图5(b)),DSSP[33]计算表明修复后二级结构含量基本可以接近模拟轨迹的水平(图5(c)).还计算了每个代表构象与同组各构象的主链RMSD,所有RMSD数值都小于2 Å (图5(a)和图5(b)),这说明二级结构的缺失只是由一些局部的偏差导致的,模型生成的大多数结构都可以通过简单修正得到合理的结果,而侧链可能存在空间冲突的情况则需要进一步改进模型来解决.

图5 T4L构象探索结果的合理性检验 (a) 使用AMBER99SB力场/OPC水模型;(b) 使用CHARMM36m力场/TIP3P水模型;(c) 修复后各代表构象的二级结构含量,参考值为模拟轨迹的平均值Fig.5.Plausibility check of T4L conformational exploration results: (a) With AMBER99SB/OPC;(b) with CHARMM36m/TIP3P;(c) secondary structure counts of each representative conformation after fixing,the reference is the average value of the simulated trajectory.
在上述流程中,闭合与打开两段模拟轨迹都被用于模型的训练.还测试了仅使用打开状态的模拟轨迹训练的情况(图6),虽然探索区域由于训练集减少而缩小,但是仍然可以覆盖包括闭合状态在内的各个晶体结构.

图6 仅从打开状态出发的T4L构象探索结果Fig.6.Results of T4L conformational exploration from the open state only.
3.2 AdK构象空间探索结果
以AdK的模拟轨迹作为训练集,进行训练以及构象空间探索.结果如图7所示,除了训练集中包含的完全关闭和完全打开状态外,还可以从中找到LID结构域单独打开(图8(a))和AMPbd结构域单独打开的结构(图8(b)).

图7 AdK的构象空间探索结果 (a) 使用AMBER99SB力场/OPC水模型;(b)使用CHARMM36m力场/TIP3P水模型Fig.7.Results of conformational space exploration of AdK: (a) With AMBER99SB/OPC;(b) with CHARMM36m/TIP3P.
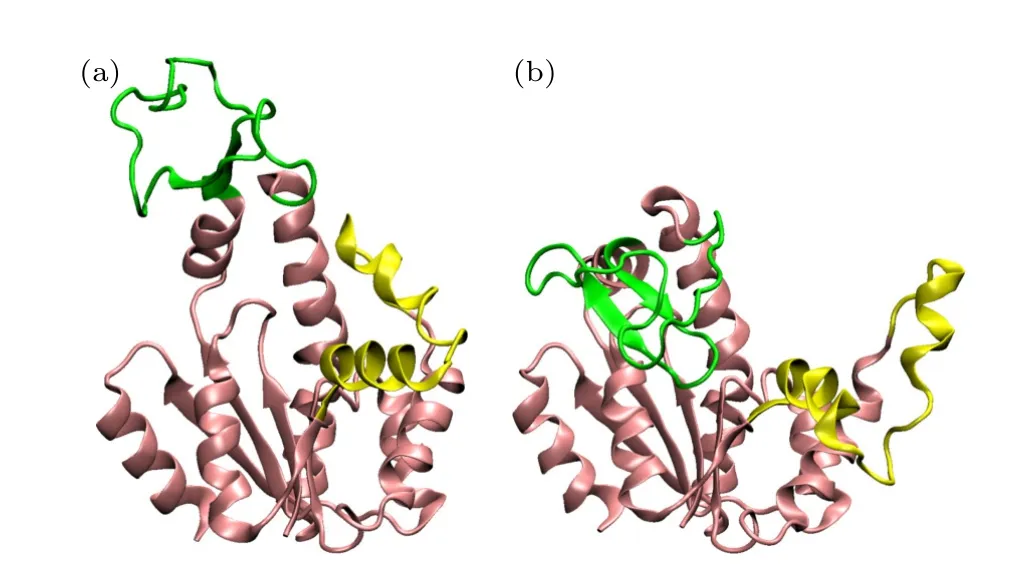
图8 探索到的不同AdK构象Fig.8.Different AdK conformations explored.
对AdK构象探索结果的合理性进行了检验,结果如图9所示.在使用CHARMM36m力场/TIP3P水模型时,修复后二级结构含量与模拟轨迹相当,而使用AMBER99SB力场/OPC水模型时,虽然也能修复到较高的水平,但与前者相比显著偏低.这表明与CHARMM36m相比,AMBER99SB力场/OPC水模型的组合使蛋白质结构更加容易发生变化,探索构象空间的范围更大,同时二级结构也会有一定的破坏,更适用于柔性较强的蛋白质分子.

图9 AdK构象探索结果的合理性检验 (a) 使用AMBER99SB力场/OPC水模型;(b)使用CHARMM36m力场/TIP3P水模型;(c) 修复后各代表构象的二级结构含量,参考值为模拟轨迹的平均值Fig.9.Plausibility check of AdK conformational exploration results: (a) With AMBER99SB/OPC;(b) with CHARMM36m/TIP3P;(c) secondary structure counts of each representative conformation after fixing,the reference is the average value of the simulated trajectory.
值得注意的是,大部分构象与其所在组的中心构象之间的RMSD较小,除少数不合理构象外,大部分RMSD较大的构象都在模拟产生的训练集中.这意味着模型产生的构象仅包含反应坐标相关的信息,而在与反应坐标正交的自由度上没有表现出差异.这是由自编码器自身的性质决定的,对于相同的输入总是会给出相同的输出,而实际上如模拟轨迹反映的一样,相同的反应坐标下,构象仍应该有一定的变化空间,这些空间是自编码器无法探索的.因此,反应坐标的选取对该模型的效果至关重要.若要解决这一问题,可以将自编码器换成变分自编码器,学习构象系综而非单个分子的特征,从而体现相同反应坐标下的差异.
以上结果是使用常规的自编码器难以获得的.将引入反应坐标监督的自编码器换成无监督的自编码器,对AdK的构象空间进行探索,结果如图10所示.自编码器需要从训练集中学习反应坐标,这在采样不足的情况下非常困难.通常情况下,自编码器只能提取两组轨迹的差异,并完成对两种状态之间的构象空间探索,但是无法探索其他区域,例如图8所示的单个结构域打开的构象.引入反应坐标作为监督的改进,使得自编码器不再需要提取反应坐标,从而可以在采样不足的情况下工作.
图10 使用普通自编码器探索AdK的构象空间Fig.10.Exploring the conformational space of AdK with a common self-encoder.
4 结论
本文对使用自编码器探索蛋白质构象空间的方法进行了改进,将监督学习引入自编码器的中间层,并使用改进后的方法对T4L和AdK的构象空间进行探索,达到了预期的效果.结果表明这一改进使该方法可以在有限采样的情况下,仅使用很少的计算资源,就可以大范围探索蛋白质的构象空间.
虽然模型只能生成构象,并不能给出构象的生物学意义以及动力学过程,但是如果对特定体系引入实验信息,就可以筛选出具有生物学意义的构象,以便进行下一步的研究.对于实验信息较少的蛋白质分子,可以直接通过模型生成有潜在研究价值的构象,然后从这些构象出发进行MD模拟,研究蛋白质分子的动态过程,进而预测可能的生物学意义.这种策略与仅依靠MD模拟的构象空间采样相比,效率更高.
在测试模型时,发现了进一步的改进空间.通过对模型生成构象的筛选和修正,可以确保构象的合理性,但同时也降低了生成构象的效率.考虑直接将对构象合理性的要求引入模型的损失函数中,从而省去筛选和修正的过程.由于模型中只有蛋白质的主链部分,有可能出现侧链不合理情况,需要对不同氨基酸残基做不同修正或在模型中使用完整的蛋白质分子.对于模型生成的构象无法表现出反应坐标之外变化的问题,可以尝试使用变分自编码器.最后,反应坐标决定了构象空间探索的方向,结合实验数据选取合适的反应坐标对模型的效果十分重要.基于这些思路,将继续对该模型进行发展和完善.
感谢中国科学技术大学超算中心张运动提供的硬件和软件技术支持.
猜你喜欢
燃烧科学与技术(2023年4期)2023-08-21
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27
中国音乐学(2022年1期)2022-05-05
轮胎工业(2020年4期)2020-03-01
物理学报(2018年10期)2018-06-14
温州大学学报(自然科学版)(2016年1期)2016-10-27
焊接(2016年8期)2016-02-27
焊接(2016年6期)2016-02-27
应用化工(2014年7期)2014-08-09
无机化学学报(2014年5期)2014-02-28
