
生物分子模拟中的机器学习方法*
2024-01-06 10:24管星悦黄恒焱彭华祺刘彦航李文飞王炜
物理学报 2023年24期
管星悦 黄恒焱 彭华祺 刘彦航 李文飞† 王炜‡
1) (南京大学物理学院,南京 210093)
2) (国科温州研究院,温州生物物理重点实验室,温州 325000)
1 引言
以分子动力学为代表的分子模拟技术在生物大分子结构与动力学研究中发挥着越来越重要的作用.常规分子模拟技术用于复杂生物分子体系时,不可避免地存在力场精度与构象采样效率瓶颈.同时,从高维分子模拟数据提取可解释的生物大分子结构与动力学特征也是一个挑战性难题.生物分子模拟技术发展的核心任务便是解决以上难题,扩展生物分子模拟的应用范围.
自从20世纪70年代McCammon等[1]首次将分子动力学模拟用于生物大分子体系以来,人们在生物分子力场发展、长程静电相互作用计算方法、增强采样与自由能计算等方面取得了多个突破[2].分子模拟技术与高性能计算机等硬件技术的协同发展使得分子模拟能够覆盖的时间尺度以超过摩尔定律的速度增加,平均每10年增加约3个数量级[3].这些进展使得人们能够直接模拟小蛋白分子毫秒时间尺度的折叠全过程[4,5],也能对固有无序蛋白(intrisically disordered protein,IDP)的构象系综进行合理的分子模拟表征[6,7],甚至能够实现对病毒颗粒、细胞质等超大分子体系进行分子模拟[8,9].目前,实验和模拟计算结合已成为生物大分子结构与动力学研究的基本范式.另一方面,对较大的分子体系,目前的生物分子模拟能够达到的空间和时间尺度与实验测量仍有一定距离,从而限制了其适用范围[10].因此,发展新的分子模拟技术,扩展分子模拟技术的适用范围,对基于生物分子模拟的基础和应用研究至关重要.
随着计算能力的提升和海量数据的积累,机器学习算法被广泛应用于基础与应用科学的各个领域.自然地,人们也将机器学习算法应用于计算生物学与生物信息学研究,如生物分子设计与结构预测、分子模拟以及分子对接等.机器学习概念诞生于20世纪50年代[11],并在曲折的发展中被多次重新理解与表述.早期的机器学习算法多是对既有建模与优化方法的重新整理与表述,如线性回归、多项式回归[12]以及k-近邻算法[13]等.尽管在早期历史中已初具雏形,目前人们广泛使用的机器学习算法,如决策树[14]、神经网络[15]、支持向量机[16]以及集成学习方法[17,18]等,大多成型于1980年后,并很快被应用于蛋白质二级结构预测[19]、蛋白结构与功能分类[20,21]以及药物筛选[22]等问题.在20世纪90年代,人们也开始将神经网络用于构建简单分子体系 (如表面吸附气体分子) 的势能面并进行分子模拟[23].在这些早期的应用中,机器学习方法往往被视为可替代的工具,且神经网络尚未表现出相对其他机器学习算法的显著优势,因此相关算法在生物分子模拟领域的应用仍非常有限.
近年来,以深度学习为代表的机器学习技术得到迅猛发展,并在多个领域展现出惊人的能力.特别是AlexNet[24]的诞生,展示了深度卷积神经网络对图像的强大识别能力,宣布深度学习革命的到来.之后出现的残差网络 (ResNet)[25]进一步推动了神经网络向深度发展,也出现了如生成对抗网络(GAN)[26]与Transformer[27]等网络架构新范式.这些新的机器学习算法开始广泛用于生物分子模拟、结构预测与设计等领域.自2017年开始,机器学习与生物分子模拟相结合的研究工作大幅增加,成为势不可挡的学科交叉趋势.这一趋势从近年来发表的相关研究论文数目的增长中可见一斑 (图1).

图1 每年结合生物分子模拟与机器学习的文献数目随年份的变化,数据来源于ScopusFig.1.Number of publications with the key words “molecular simulations” and “machine learning” published per year as a function of years.Data were taken from Scopus.
机器学习与生物分子模拟的结合为推进分子生物物理学研究提供了新的机会.例如,利用机器学习技术能够设计更准确的分子力场,开发更高效灵活的增强采样算法,发展更具普适性的复杂生物分子体系的结构与动力学预测算法,并辅助药物分子的设计.这一重要的交叉领域正在高速发展并持续产生具有突破性进展的研究成果[28-35].因此对该领域的发展进行回顾与综述尤为重要.关于机器学习在生物大分子结构预测与设计方面的进展,已有非常全面的综述可供参考[36-40],本文不再过多讨论.在机器学习与生物分子模拟交叉领域,也有学者从不同角度进行了综述[41-44].例如,Ramanathand等[42]在其综述论文中介绍了使用机器学习技术表征IDP系综以及进行多尺度模拟的方法,并提出将模拟数据集与实验拟合的重要性及策略;Noé等[43]详细介绍了机器学习算法在帮助解决生物分子模拟重要挑战中发挥的作用,并探讨了将物理学原理融入机器学习算法的必要性及相关方法;Wang等[44]详细总结了利用机器学习算法分析分子动力学模拟轨迹的方法,以及利用机器学习与相关数据驱动方法进行增强采样的方案.本文将在此基础上,结合该领域的最新进展,从生物分子力场构建、反应坐标的选取与增强采样、分子模拟数据处理等方面对机器学习与分子模拟交叉领域的代表性工作进行综述.生物物理智识与机器学习技术迭代的融合已成为人们探索生命原理的有力手段,而结合机器学习算法的生物分子模拟是借助神经网络的强大表达性与拟合能力分析复杂生命运动密码的重要实践.期望本文对该领域的综述有助于读者综合了解机器学习算法在生物分子模拟中的重要应用,共同思考和探索基于机器学习算法解决生物分子模拟领域关键难题的可能途径.
2 基于机器学习算法的生物分子力场构建
2.1 势能面与分子力场拟合
在生物分子模拟中,精度和效率通常难以兼得.不同的问题在精度和效率上有不同的偏重与要求,因此需要针对性地选择能够平衡精度与效率要求的折中方案.计算化学领域的“金标准”CCSD(T)方法能达到约1 kcal/mol的化学精度,但代价是计算效率低,通常适用于小体系的单点能计算.基于密度泛函理论 (DFT) 和Born-Oppenheimer绝热近似的方法在精度上作出妥协,从而提升了计算效率,能够将计算体系大小提升到数百个原子以上的规模.但是,对于绝大多数的生物大分子,计算体系通常包含上万个原子,并涉及微秒以上的时间尺度,因此进一步提升生物分子模拟的计算效率对扩展其应用范围十分关键.分子力场模型通过参数化力场的方式在原子坐标水平近似地描述绝热能量面,从而大幅提升计算模拟效率.这种逐级近似的框架之下,如何在提升计算模拟效率的同时尽可能减小精度的损失,成为构建分子力场的核心问题.全原子水平的分子力场可以看作是基于原子坐标和原子类型的高维空间上的多元函数.传统分子力场多使用基于经验的结构项和以单体、两体势表示的非键相互作用项的参数化方案[45-47].这种预先设定的具体力场函数形式不可避免地对力场精度带来限制.尽管人们可以通过进一步引入极化和多体效应等物理机制来提升参数化方案的表达能力[48,49],但在精度上与DFT方法仍有较大差距.深度学习算法提供了一种表达能力强大的参数化方案(图2),可以降低对预设力场函数形式的依赖,因此原则上可以提升对分子力场的描述精度.需要注意的是,深度学习算法更强的参数化表达能力,需要由充足的计算能力和训练数据来作为支撑.近年来,计算能力与数据规模已经可以支持用于训练具有足够强表达能力的深度神经网络,因此使用深度学习方法构建生物分子力场,从而实现分子力场精度突破的条件已经成熟,且在此问题上已取得重要进展[50-55].

图2 神经网络用于生物分子构象能量面及力场的拟合Fig.2.Schematic diagram for representing the biomolecular force field by a neural network.
机器学习算法用于生物分子力场拟合的一个典型例子是Zhang等[51,56]在2018年发表的Dee PMD工作.DeePMD使用原子尺度的构象坐标以及量子力学精度的能量信息作为数据集,将系统构象映射至其对应的能量与力(受益于神经网络组件的求导能力).给定系统构象坐标,可以通过网络的前向传播代替复杂的DFT计算,直接得到原子受力,从而在尽量保留DFT精度的前提下实现高效率分子动力学模拟.DeePMD的网络架构本身是深度前馈网络,由多个全连接网络的输出求和得到总能量.DeePMD使用分子构型的相对坐标来保证网络的输出不依赖于生物分子体系的平移与旋转变换.值得一提的是,DeePMD可以对接LAMMPS,Gromacs等传统分子动力学模拟软件,便于使用.
为了在神经网络训练中保持分子构型平移与旋转对称性,除使用相对坐标(或单个分子体系的内坐标)外,另一类方法是使用Behler与Parrinello[57]在2007年提出的对称函数方法.对称函数方法将系统中每一个原子依次视为中心原子,计算其与附近原子的距离、夹角,得到对称函数值,并作为神经网络的输入特征量.例如,Artrith与Urban[58]发展的Aenet神经网络模型以及Smith等[59]发展的ANI-1神经网络模型均使用了该对称函数方法,并成功用于体相TiO2等材料系统和有机物小分子系统的力场拟合.Fan等[60]在基于进化策略算法构建用于原子模拟的机器学习势时也采用了类似的方法.该对称函数方法规避了笛卡尔坐标与内坐标的相互转换,从而提升深度网络的参数表达能力和训练效率.
以上DeepMD,Aenet,以及ANI-1均采用了深度前馈网络构架.随着卷积神经网络 (CNN) 展示出其对图像特征提取与识别的强大能力并在机器学习领域带来革命,人们也尝试使用CNN处理图像的范式来处理分子构型并映射到能量面或力场.特别是残差网络构架的引入,使得人们可以在避免过拟合的前提下,构建足够深度的CNN网络,以增强其拟合效果.一个代表性的例子是Schütt等[50]发展的SchNet.SchNet以残差卷积网络实现对分子构型特征的提取.不同于处理图像数据使用的网格状离散滤波器,为了保证能量面的光滑性与精确性,SchNet采用了连续滤波器.相对于深度前馈网络,基于CNN架构的SchNet能够显著提升在量子化学精度数据集QM9 (包含有机小分子的构型、能量等)的预测精度,也在分子动力学数据集MD17[61]上有更好的表现.
尽管CNN可以提取局域而抽象的特征,且相较于全连接神经网络在避免出现过拟合方面表现出色,但CNN最擅长的领域仍是处理规整的图像等数据.对于空间不规则且以共价链接为重要特征的分子构型,图(graph)是一种更为自然的表示.分子构型的图描述天然地拥有平移和旋转不变性,并且允许将距离、化学键等连接信息作为“边”数据存入图网络.因为这些优点,人们也尝试使用图神经网络来学习拟合分子力场.Park等[53]于2021年发表的GNNFF基于结合有向图与消息传递 (message passing)的深度神经网络框架[62],构建了神经网络分子力场模型,对有机小分子受力的预测精度超过SchNet.Wang等[63]在同年发表的sGNN,考虑了不同类型相互作用在空间尺度上的差异,对聚合物分子的主链共价作用和非键相互作用能量项分开建模,在空间尺度扩展性与对不同模拟体系的可迁移性方面表现良好.
2.2 粗粒化力场构建
相对于DFT等量子化学方法,基于分子力场的全原子分子动力学模型极大地扩展了计算模拟方法能够研究的生物分子体系的空间和时间尺度.目前,人们已经能够实现对较小蛋白体系的完整折叠过程进行全原子分子动力学模拟.另外,通过结合增强采样算法,可以实现对较大生物分子体系构象变化的全原子分子动力学模拟和自由能计算.然而,对于更大的生物分子系统,如分子马达、核糖体、病毒颗粒以及染色质体系等,通常包含百万以上原子个数,并涉及毫秒以上时间尺度的动力学过程,远超出全原子分子动力学模拟能够达到的时间和空间尺度范围.为了突破全原子分子动力学模拟的计算效率瓶颈,人们通常采用粗粒化的近似方法[64].在粗粒化模型中,将多个原子映射为1个虚拟粒子,从而很大程度上降低了体系的自由度数,实现分子模拟效率的提升.然而,由于采用了虚拟粒子近似,构建具有合理精度的粗粒化分子力场是一个极具挑战性的难题.已有的粗粒化模型的力场参数主要通过“自下而上”和“自上而下”两种策略来优化得到.
“自下而上”策略的基本思路是基于高精度力场模型的计算结果来确定粗粒化力场参数,主要方法有玻尔兹曼反演法 (Boltzmann inversion method)[65]、力匹配法 (force matching)[66]、涨落匹配法(fluctuating matching)[67]以及能量分解法 (energy decomposition)[68,69]等.例如,玻尔兹曼反演法主要通过全原子分子动力学模拟得到的径向分布函数 (radial distribution function) 来提取粗粒化层次的有效相互作用参数;而力匹配法的优化目标则是使粗粒化粒子的受力与其在高精度力场中对应粒子的受力尽可能一致.需要注意的是,由于粗粒化近似,粗粒化粒子所代表的原子体系的自由度被冻结,粗粒化力场需要包含所冻结自由度构象熵对能量面的贡献,因此是一种平均力势 (potential of mean force).
以上基于“自下而上”方案构建粗粒化力场的策略与前述基于DFT计算结果拟合全原子力场的思路相类似,都希望基于低精度模型拟合更高精度的数据 (能量或力),从而在提升计算效率的同时,尽可能保留足够的精确度.不同的是,从量子力学模型到全原子分子力场模型,由于原子自由度数目维持不变,因此分子力场不涉及构象熵的贡献,原子尺度力场的拟合可以直接使用能量或力作为目标;而在构建粗粒化分子力场模型时,需要在一定程度上体现被冻结自由度的熵效应,因此对分子构象的采样具有更高的要求,将力作为目标拟合力场参数是更常用的方法.另外,构建全原子力场模型的相关算法和构架,如神经网络架构、体现平移与旋转对称性的结构特征提取方法、激活函数的选择等,可以自然地迁移到基于力匹配的粗粒化力场拟合.近年来,基于深度学习构建粗粒化分子模型的工作越来越多地见诸于发表的论文中[34,52,70-74].例如,DeePMD团队同时开发出与DeePMD具有相似网络架构与结构特征提取策略的深度学习粗粒化力场方案——DeePCG[52].其中力场参数的提取使用了力匹配法和逐级拟合的办法.同样是基于前馈神经网络架构和力匹配方法,Wang等[70]在2019年开发了CGNet,并展示了用于丙氨酸二肽与多肽链的粗粒化模拟结果,能够很好地重现作为参考的全原子模拟得到的自由能面及其他统计性质.
以上例子均采用了基于“自下而上”思路的力匹配法作为粗粒化力场拟合方案.与其相对应的“自上而下”的思路追求粗粒化力场模拟结果与实验约束或高精度模型得到的宏观性质的相容性.然而,因为每一步优化都需要在当前参数下得到模拟轨迹并进行反向传播,自上而下的方法通常会给训练带来较大的计算负担,对拟合目标与参数优化方案的选择具有更高要求[75,76].近期Clementi和Noé等[34,71]提出了以flow-matching为例的一类新方法: 将标准化流 (normalizing flow,NF) 或去噪扩散模型 (denoising diffusion probabilistic model)等生成模型与力匹配法相结合,先利用高精度数据训练粗粒化构象的生成模型,再从这种生成模型中提取粗粒化力场.这些新的方法将生成模型描述的粗粒化构象偏好视作一种平衡采样,从而与力场产生联系.其他的生成模型,如变分自编码器 (variational auto-encoder,VAE)[72]和使用对抗训练思想的VADE[73]同样可以被用于描述粗粒化坐标下的构象分布.
另外,在基于Cα的蛋白质粗粒化模型中,由于侧链原子位置信息的缺失,无法准确地体现蛋白质分子的表面积、静电势分布等蛋白质分子的基本性质.但是这些信息对理解蛋白质分子的结构组装、构象动力学以及分子识别等过程至关重要.因此,如何在粗粒化模型框架下准确地计算蛋白质分子的表面积、静电势等蛋白质分子的基本性质是一个重要的技术挑战.基于深度神经网络的机器学习算法为解决这一问题提供了一个可行的方案.例如,本文作者在最近的工作中,构建了一套深度学习网络DeepCGSA,能够基于粗粒化模型结构高精度地估算蛋白质、核酸等生物大分子的溶剂可及性表面积(图3)[74].尝试将类似的方法用于针对粗粒化蛋白质结构的静电势分布与pKa值的预测也取得了很好的效果.

图3 基于粗粒化结构的蛋白残基溶剂可及性表面积(SASA)计算.左图: 蛋白分子(protein G,PDB code:1pgb)的全原子结构图与粗粒化结构图;右图: 使用DeepCGSA由粗粒化结构计算得到的SASA与参考值的对比.其中参考值使用Shrake-Rupley算法由全原子结构计算得到[77].DeepCGSA能够基于粗粒化结构给出接近参考值的SASA计算结果Fig.3.SASA estimation based on coarse-grained protein structure.Left: All-atom structure and coarse-grained structure of protein G (PDB code: 1 pgb).Right: Correlation plot between the SASA values from DeepCGSA based on one-bead coarse-grained structure and the reference values by Shrake-Rupley algorithm based on all-atom structure.The DeepCGSA can well reproduce the SASA values based on coarse-grained structure.
3 基于机器学习算法的分子模拟增强采样与数据处理
由于生物大分子具有庞大的自由度数和复杂的能量面特征,全原子水平的分子模拟通常会遇到采样困难.特别是在计算各种平衡统计性质时,需要分子模拟的采样尽可能遍历重要的构象空间,并在给定的系综条件下达到平衡.尽管上述粗粒化模型提供了一种解决采样困难的有效方案,但粗粒化近似不可避免地导致计算精度的损失.特别是当特异性的氢键、盐桥等原子层次的相互作用起到主导作用时,粗粒化模型通常无法显式地体现这类特异性相互作用特征,从而限制了其应用范围.因此,发展增强采样算法是解决分子模拟采样困难的另一有效方案.基于统计物理原理,人们已经发展出多个有效的增强采样算法,并广泛应用于生物大分子体系的蒙特卡罗模拟和分子动力学模拟[78-88].目前常见的增强采样算法有伞形抽样(umbrella sampling)[78]、副本交换分子动力学 (replica exchange molecular dynamics)[79]、元动力学 (metadynamics)[80]、加速分子动力学 (accelerated molecular dynamics)[81]以及温度积分增强抽样方法 (integrated tempering sampling,ITS)[82]等.这些增强采样算法多已通过外部插件(如PLUMED[83]) 或直接整合到成熟的分子动力学模拟软件.另外,人们也发展了适用于研究构象转变路径的增强采样算法,如String方法[84]与Transition path sampling方法[85]等.最近,人们将机器学习算法用于生物分子模拟的增强采样,并取得了显著效果,甚至还可以利用机器学习算法,基于有限的构象采样数据实现高维自由能面的构建[89,90].
3.1 基于机器学习算法提取反应坐标
常用的增强采样算法可分为两类: 依赖反应坐标的增强采样算法和不依赖反应坐标的增强采样算法.例如,伞形抽样、元动力学等增强采样算法依赖于预先定义的反应坐标,这类算法的基本策略通常是沿预先定义的反应坐标方向添加偏置势,从而避免在沿反应坐标的局部势阱中重复采样.因此,预先定义的反应坐标需对应所关注的生物分子体系最重要的运动方向,而垂直于反应坐标方向的动力学具有更快的时间尺度.然而,定义合适的反应坐标本身就是一项极具挑战性的任务.通常情况下,反应坐标主要基于物理直觉来选取,而机器学习等数据驱动的降维方法为反应坐标的选取给出了一个更为理性和可操作的方案.
常规的不使用神经网络的数据驱动降维方法主要基于如下思想设计: 在降维前后的空间里,尽可能维持数据的某种结构信息不变.这种“结构信息”可以分为全局信息和局域信息两类.早在20世纪初就被开发的主元分析算法PCA,是一种典型的致力于维持全局结构信息的算法[91].PCA将高维数据点相对于几何中心的欧式距离平方和视作需要保留的“结构信息”,在通过线性变化降维过程中最小化该结构信息的损失,并找到承担最大运动信息变化的反应坐标.PCA方法的缺陷也在于此: 基于全局的欧式距离衡量信息并非总是一个合理的预设;且PCA要求降维至超平面,就只允许对数据做全局的线性变换,很多时候这是一个过强的假设.
更一般地,可以假设高维数据分布在一个黎曼流形(或是几支黎曼流形)上.此时欧式距离只适用于描述数据点的局域结构,即可以构建起离散数据点的近邻图,而全局结构可视为由这些近邻图组合而成.基于这一思想,Isomap算法[92]和Diffussion Map算法[93]分别用测地线距离和模拟扩散距离衡量数据点的间距,并希望降维映射前后这些距离尽量保持不变,从而将流形“展平”以实现降维.将Isomap与Diffusion Map用于分子模拟数据分析,可以找到非线性地依赖于高维数据的反应坐标[94-96].
在基于局域结构信息的降维方法中,2008年提出的t-SNE算法具有突出的表现[97].t-SNE对数据点间的相似性做非线性变换,使得降维过程中主要维护局部团簇 (cluster) 中两点相似性的分布不变,而对相似性低的数据点的位置关系几乎没有约束.因此,t-SNE的降维尽量维持了数据点基于相似性簇团的内部结构,而对簇团间的距离朝向则几乎没有要求,从而带来了降维结果的随机性.t-SNE使用梯度下降优化低维空间数据点的位置,通常这是一个非凸优化,每次得到的结果会有所差别.相比于2002年提出的SNE算法[98],t-SNE构建对称的损失函数以代替SNE中不对称的K-L散度,简化了基于梯度的优化过程;同时t-SNE以更为长尾的t-分布建立低维空间距离向概率的映射,以更好应对高维数据点嵌入低维空间导致的拥挤问题.图4给出了使用PCA,t-SNE以及UMAP对粗粒化分子动力学得到的蛋白折叠轨迹[99]进行降维的效果对比: 相比于PCA,t-SNE和UMAP能更好地区分折叠态和解折叠态的结构.在分子模拟中,基于t-SNE的降维算法已被广泛应用于反应坐标的定义与高维动力学轨迹的可视化[100-102].除t-SNE外,基于局域结构信息的降维方法还有: 维持局域线性关系的LLE (locally linear embedding)[103]、维持局域邻近图的Laplacian Eigenmaps[104]、最小化局域曲率的Hessian LLE[105]等,然而它们在分子模拟领域得到的关注和应用远不如t-SNE.2018年McInnes等[106]提出的UMAP降维算法采用了与t-SNE类似的、基于邻近图提取簇团信息的策略,并同样用梯度下降方法优化得到低维嵌入.不同的是,相比于围绕着“点”进行的t-SNE,UMAP采用了以“边”为中心的优化策略,使用交叉熵作为优化目标,将边存在的概率映射为低维空间的距离.在生物分子模拟中,UMAP常被用于基因组、染色质和单细胞转录谱等数据[107,108].在单细胞转录谱数据集与蛋白质动力学轨迹数据上的比较研究[109-111]均表明: UMAP具有不逊色于t-SNE的降维效果,但是在计算成本上远低于t-SNE,对大规模的数据有良好的扩展性,这与UMAP原始论文中指出其计算复杂度约为N1.14一致[106].
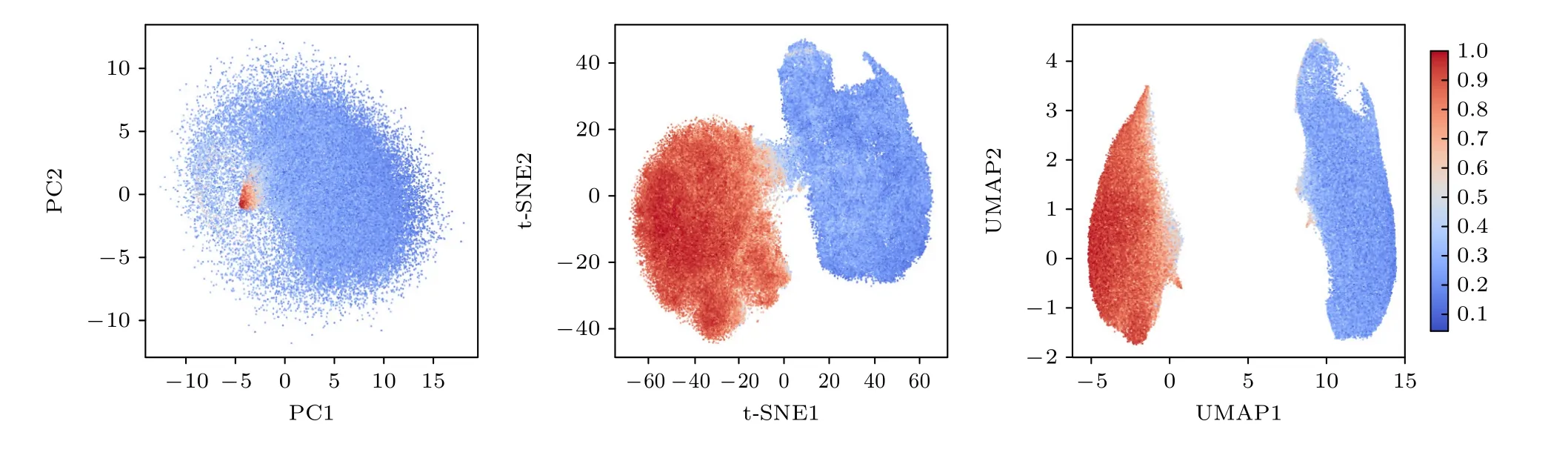
图4 用PCA (左)、t-SNE (中)和UMAP(右)对蛋白分子Protein G的基于粗粒化分子动力学的模拟轨迹[99] 降维效果对比.蓝色到红色对应表征蛋白折叠程度的Q值;Q=1 (红色)为完全折叠结构,Q=0 (蓝色)为完全解折叠结构Fig.4.Projection of the sampled snapshots of the coarse-grained molecular dynamics simulations for protein G [99] along the reaction coordinates constructed by PCA (left),t-SNE (middle),and UMAP (right),respectively.t-SNE and UMAP perform better than PCA in distinguishing the folded and unfolded structures.Colors from blue to red represent the structures with increasing folding extent: blue,fully unfolded;red,fully folded.
如果认为降维算法的关键问题在于对信息的选择与度量,那么以上非神经网络的机器学习降维算法都是通过引入某种预设 (或主观判断) 来解决此问题,也因此降低了对降维变换的表达能力.借助于具有强大表达能力的神经网络,可以期待构建更有效的降维算法.
在2013年被开发的VAE,通过巧妙地设计神经网络架构,将原始数据通过编码器降维得到隐变量,再通过解码器升维,生成与原始数据同维度的高维数据[112].如果生成数据具有和原始数据几乎相同的分布,则说明编码过程 (即降维过程) 几乎没有造成信息损失,低维的隐变量具有与原始数据相近的表达能力.就训练过程而言,VAE通过优化编码器和解码器参数,以最小化生成数据与原始数据分布上的差异.其中,隐变量的“信息”通过复现原始数据分布的能力衡量.相比于以上非神经网络的降维算法中预设信息为数据集上的某种结构的做法,VAE衡量信息的方式更具一般性与整体性.对于生物分子模拟系统,这一优势将有利于VAE通过降维找到整体性的反应坐标;而编码器、解码器所基于的深度神经网络架构保证了VAE强大的表达能力,降低了模型对预设信息的依赖,有利于增强降维的有效性.因此VAE常被用于生物分子模拟反应坐标的提取.另外,VAE寻找反应坐标的思路同样可以用于粗粒化模型的建立[72]、反应路径搜索[113]、甚至是药物分子设计[114]等任务.
3.2 基于机器学习算法的增强采样
3.2.1 非生成模型
机器学习算法不仅可以用于寻找合适的反应坐标,还可以直接用于辅助分子模拟采样.例如,在利用Metadynamics方法进行增强采样和自由能计算时,需要在分子体系的固有能量面添加一定形状的高斯形偏置势[115],而确定高斯形偏置势的参数及其变化规律非常关键,直接影响采样效率.过强的高斯形偏置势可能会导致采样进入非物理的区域,而过弱的高斯形偏置势又难以遍历感兴趣的构象空间区域.2019年,Bonati等[116]通过结合神经网络与变分增强采样思路,灵活地以变分形式在增强采样模拟过程中自适应地更新偏置势,使得反应坐标的实际分布能够逼近目标分布.相较于常规的Metadynamics方法,在灵活性、高效性与准确性方面得到了提升.
另一个使用神经网络给出偏置势用以增强采样的例子是Zhang等[117]提出的TALOS (targeted adversarial learning optimized Ssampling).类似生成对抗网络GAN的思想 (见下方关于生成模型的描述),TALOS使用Wasserstein距离衡量真实分子模拟引擎生成的构型分布与目标构型分布的差异,将此距离的计算转化为对一个判别器网络的优化问题.TALOS的训练同样类似于GAN: 对每个偏置势,通过优化判别器网络计算两分布Wasserstein距离的近似值,以之作为两分布差异的数值衡量;最小化此差异以优化偏置势,从而使偏置势下模拟产生的构型分布尽可能接近目标分布.
3.2.2 生成模型
在以上例子中,机器学习算法仅被用作提供构造反应坐标或设置偏置势的手段,即增强采样的辅助工具,并没有直接采样生成分子构型.近年来生成模型的发展,使得人们可以借助神经网络直接生成生物分子构型,这为发展新的增强采样算法提供了新思路.目前广泛使用的生成模型主要包括VAE[112],GAN[26]和标准化流模型[118]等(图5).前面已经提到VAE通过编码器-解码器对原始数据进行降维再升维后,使生成数据尽可能与原数据保持相似,这一相似性的要求体现在损失函数中的K-L散度.但实际上VAE的损失函数是两部分的拮抗,另一部分则是希望低维空间的隐变量尽可能接近高斯分布,于是解码器并不是编码器的逆变换,而是在拟合低维空间高斯分布参数后进行的采样.生成对抗网络GAN则是用一个分类器来判定生成器生成的结构是否合理 (被生成的分子构型与原数据是否相似这一判据由分类器训练得到),那么由生成器生成并被分类器所选择的分子构型便可以作为采样结果.标准化流模型则是用一系列变换在两种分布间搭建可逆变换.

图5 不同生成模型的网络架构.从左至右分别对应变分自编码器、生成对抗网络与标准化流.即便目标同为生成符合某种分布的数据,三种网络使用了不同的架构与方法.变分自编码器将数据降维至低维空间后,在低维空间采样并再次变换至高维空间;生成对抗网络则通过生成器与分类器之间的互相对抗而使生成器生成的结果符合目标分布;标准化流则是在目标分布与简单易采样的分布 (如高斯分布) 之间建立直接且可逆的映射Fig.5.Network architecture of different generative models:Variational autoencoder (VAE,left),generative adversarial network (GAN,middle),and normalizing flow (NF,right).Three networks have different architectures.VAE first reduces data to a low-dimensional space,samples in the lowdimensional space,and then transforms back to a high-dimensional space.GAN generates target distribution by combining a generator and the discriminator.Normalizing flow model establishes a direct and reversible mapping between the target distribution and a simple and easy-tosample distribution (such as Gaussian distribution).
用神经网络本身作为采样核心的一个代表性例子是Noé等[28]2019年发表于Science的Boltzmann Generator.该工作完全展示了标准化流模型作为数学优美的可逆生成模型在两种分布之间建立联系的能力.真实的分子构型的分布符合玻尔兹曼分布,通常这一分布难以直接采样.Boltzmann Generator的标准化流模型通过构建可逆的坐标变换,将简单且容易采样的高斯分布映射到玻尔兹曼分布,从而实现满足玻尔兹曼分布的采样.
假设变量x取自高斯分布,概率密度为q(x),z代表生物大分子结构的原子坐标,概率密度为r(z),应当符合玻尔兹曼分布.需要注意的是,即使通过训练建立起变量之间的映射函数z=f(x),变量z的概率密度并不是简单的r(z)=r(f(x)),而是需要考虑体积元变化:
在保持变换可逆的同时计算出雅可比因子是一个难点.而标准化流模型使用了如下精巧设计: 用一系列小变换一步步将x变换到z,并且每一个小变换都是可逆且雅可比因子易计算的.每一步变换将x划分为两部分x=(x1,x2),:
该变换的可逆性也显而易见.如果变换使得雅可比矩阵呈三角矩阵,则可非常容易地计算得到雅可比因子:
在Boltzmann Generator的实际训练中,可以选择多种训练方式: 1)将实际轨迹作为玻尔兹曼分布端的输入z,并通过训练将经变换输出的x优化为高斯分布;2)将根据高斯分布采样得到的x变换得到构型z,并根据分子力场计算其能量,通过训练使z的分布优化到玻尔兹曼分布;3)结合前两种方法训练;4)在前三种选项的基础上加入额外的依赖于反应坐标的损失项,使得变换后的分子构型尽可能在反应坐标空间均匀分布 (类似Metadynamics的思想),此后再进行reweighting操作,得到正确的分布.在将Boltzmann Generator用于BPTI蛋白的构象采样时,成功得到了其“X”态到开放的“O”态之间的构象转变,即使这种转变的过渡态并不存在于训练集中,展现了Boltzmann Generator在用于生物分子构象采样的强大能力.
另外一类常见的增强采样算法采用了强化学习方法.强化学习使用奖惩机制,在不同的环境条件下强化学习器采取不同的动作时将给出一定的奖励或惩罚,而训练的目标是使得强化学习的动作能够将奖励最大化.Shamsi等[119]提出了基于强化学习的REAP算法,将奖惩机制与分子构象空间的探索绑定在一起,寻找最利于在构象空间扩散的反应坐标.该方法用于丙氨酸二肽和Src激酶体系时展示了出色的增强采样的效果.基于类似的思想,人们也可以基于强化学习,在沿所设定反应坐标采样的不确定度(uncertainty)上施加奖惩来鼓励体系在未遍历的构象区域采样(在已经遍历的方向上反应坐标的不确定度较低),因此能对增强采样模拟施加一个自适应的灵活偏置势[120],达到增强采样的效果.
综上,机器学习在增强采样领域表现出强大的功能和应用前景,既可以在传统增强采样算法框架下通过构建反应坐标发挥作用,也可以通过自适应的方式提供更高效灵活的偏置势,还可以直接利用生成模型作为采样核心.随着新的机器学习算法的开发,将机器学习用于辅助生物分子模拟增强采样是未来生物分子模拟领域的重要课题.
3.3 基于机器学习算法的生物分子模拟数据处理
生物分子模拟通常在高维空间中进行,所得到的分子模拟轨迹包含了丰富的结构与动力学信息,如何从这些高维的分子模拟轨迹提取出可解释的热力学与动力学数据,并实现与实验结果的定量比较是分子模拟领域的另一个挑战性难题.分子动力学模拟数据处理主要包括以下几个方面: 高维分子模拟数据特征提取、分子模拟轨迹降维与反应坐标构建、分子模拟微观状态粗粒化与马尔可夫状态模型构建,以及低维自由能面构建等.显然,适合于处理复杂数据的各类机器学习算法在生物分子模拟数据处理中扮演着越来越重要的角色.事实上,前述关于增强采样算法部分介绍的基于机器学习的反应坐标构建是机器学习用于分子模拟数据处理的重要方面.除此之外,人们也发展了深度网络模型,用于提取生物分子体系的动力学与自由能信息.例如,Mardt等[121]设计了VAMPNet,能够端到端地直接实现从分子模拟数据轨迹得到马尔科夫状态模型(Markov state model)的映射.以马尔可夫过程的变分法(VAMP)为基础,深度网络用于表达特征变换的形式,通过变换后的特征空间内近似得到弛豫时间τ范围内的状态转移矩阵,从而用于提取生物分子的动力学信息.另外,Schneider等[90]通过训练神经网络,实现了高维自由能的计算以及典型系综平均性质的计算.
4 总结与展望
本文对机器学习方法在生物分子模拟领域的应用进行了综述.借助其突出的特征提取和参数拟合能力,机器学习方法(特别是神经网络算法)在全原子/粗粒化分子力场构建、分子模拟数据降维与反应坐标提取、以及生物分子构象采样等方面已经开始发挥重要作用.随着以深度神经网络为代表的机器学习算法的迭代更新,结合机器学习算法的生物分子模拟技术将成为人们在分子层次探索生命原理的重要研究范式.需要指出的是,目前机器学习算法大多作为辅助工具在生物分子模拟中发挥作用.即使整合了机器学习算法,对较大的生物分子体系能够达到的分子模拟时间尺度仍与真实生物学相关时间尺度有较大差距.完全解决生物分子模拟精度与效率瓶颈,实现生物分子模拟与实验测量的定量比较,需要在分子模拟的理论框架与算法方面同时进行探索.近年来,整合全原子模型和粗粒化模型的多尺度生物分子模拟技术越来越受到人们的重视[67,122-124],是解决生物分子模拟精度与效率瓶颈的一个值得重点尝试的思路.神经网络等机器学习算法的发展将成为进一步推动多尺度分子模拟技术发展的新突破口.
尽管本文将机器学习用于生物分子模拟的工作分为力场构建、增强采样以及数据处理等不同的主题来进行综述,近年来突破性的工作通常打破了主题分类的边界,并依赖于多个步骤的集成耦合.因此,实现机器学习在生物分子模拟多方面的融合应用,需要开发能够集成机器学习算法与生物分子模拟的软件平台.例如机器学习与生物物理交叉领域代表工作——AlphaFold2与ESM大语言模型,均得益于对多模态数据与算法的集成整合能力[30,125].国内在相关领域的集成软件平台开发方面也取得了很大进展.由深势科技开发的RiDYMO平台集成了神经网络、分子动力学引擎、增强采样方法,不仅能进行分子动力学模拟,分析蛋白质构象空间、还能探索药物结合位点并计算药效相关动力学参数,适合药物的设计与开发工作[126].北京大学与华为等团队开发的MindSPONGE[127]在华为昇思MindSpore框架下整合了多种分子模拟、结构预测设计以及全面的神经网络支持.这些集成平台将降低新算法的开发和使用门槛,促进生物分子模拟技术的应用范围.
关于机器学习与生物分子模拟融合应用的研究进展给我们带来一个重要的启示: 生物物理智识与机器学习发展是相辅相成的.例如,AlphaFold2的架构借鉴了由序列比对得到的共进化信息,而AlphaFold2的成功又是机器学习推进生物分子结构预测领域的代表例子.生物分子模拟与神经网络结合的需求也同样在推进机器学习领域的发展.SchNet为了拟合光滑连续力场而在卷积神经网络架构下提出的连续滤波器,可以被推广到其他机器学习任务情景;而主要受分子结构拓扑相关研究驱动而发展的图神经网络,也被推广到诸如社交网络等应用情景中.机器学习架构的每一次突破性进展都会为生物分子研究领域带来难以估量的灵感与启发.如何借助神经网络的成功进一步反哺生物物理智识与经验将是未来生物物理与人工智能交叉领域的重点研究课题.
猜你喜欢
燃烧科学与技术(2023年4期)2023-08-21
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27
车主之友(2022年4期)2022-08-27
中国音乐学(2022年1期)2022-05-05
海峡姐妹(2019年12期)2020-01-14
广西农学报(2019年4期)2019-11-26
物理学报(2018年10期)2018-06-14
温州职业技术学院学报(2014年3期)2014-03-11
计算物理(2014年1期)2014-03-11
燕山大学学报(2014年1期)2014-03-11
