
基于不同势函数的正构烷烃物理性质分子动力学模拟
2023-08-21 06:37:18陈飞龙张延志何志霞
燃烧科学与技术 2023年4期
陈飞龙,张延志,何志霞
基于不同势函数的正构烷烃物理性质分子动力学模拟
陈飞龙1,张延志1,何志霞2
(1. 大连理工大学能源与动力学院,大连 116024;2. 江苏大学能源研究院,镇江 212013)
采用分子动力学模拟详细比较了4种常用势函数对正构烷烃物理性质预测精度的影响,提出了一种新的修正力场并对其进行广泛的验证,验证主要包括饱和两相密度、饱和蒸气压和自扩散系数等热物理和输运性质.之后,将改进力场应用于二元组分燃料热物性研究中,探究了不同混合比例条件下通过每种组分加权平均的方法估算混合物饱和热物性的准确度.研究发现,改进力场对正庚烷和正十二烷物理性质的预测与实验值较为吻合;在温度较低、正庚烷摩尔分数较高或者温度较高、正庚烷摩尔分数较低的工况下,通过加权平均得到的饱和物性值较为准确.
分子动力学模拟;物理性质;势函数;二元组分
步入全球现代化的21世纪以来,化石燃料具有潜在的能源短缺的危机,燃烧化石燃料(主要是碳氢燃料)而产生的二氧化碳是加快全球变暖的温室气体的主要来源之一.面对能源短缺和环境恶化的双重危机,节约能源和清洁低碳化发展已成为全球共识.除了发展可再生能源和探索替代能源之外,提高碳氢燃料的燃烧利用效率至关重要.实现这一目标的基础和前提是对碳氢化合物各项物理性质的定量估计,而对于燃料在一些极端工况如高温高压的环境条件下或者是不同组分混合物热力学以及输运性质的实验数据比较匮乏,且实验成本较高,因此用理论预测的手段来补充很有必要.
状态方程是预测流体热物性的有效手段,但是先决条件是需要进行各种假设、需要提供经验参数以及混合规则等,存在对于不同环境不同组分的适用性问题,同时计算效率和精度等问题也同样需要考虑.为了避免传统数值模拟和实验所遇到的问题,分子动力学(molecular dynamics,MD)模拟技术近年来被越来越地多应用于流体性质的研究.MD模拟的优势是不需要引入对所研究物理过程的任何假设,只要有能正确描述物质粒子间作用力的势函数,适用于对各项热力学以及输运性质的预测.
MD计算中,势函数作为描述粒子间作用力近似数学表达方法的选取极为关键.碳氢化合物的常用分子模型主要分为全原子模型和联合原子模型,前者视每个原子为一个位点,后者将每个甲基CH3和亚甲基CH2基团分别视为一个联合原子来简化计算.最早出现的有Jorgensen等[1-4]提出的OPLS-AA和OPLS-UA力场,广泛应用于蛋白质、烃类和醇类等复杂的大分子物质.之后Siepmann等[5-6]提出的SKS力场,更适用于模拟链长较短的烷烃.除此之外,又相继出现了Martin等[7]通过拟合烷烃的临界温度和饱和液相密度的实验数据提出的TraPPE-UA力场以及Nath等[8]提出的对短长链烷烃都适用的NERD力场.邓磊[9]比较了OPLS-AA力场以及一种联合原子力场对正庚烷液滴的蒸发特性的影响,结果表明,在采用截断半径法的计算下所选用的联合原子力场比OPLS-AA力场对正庚烷粒子间作用力的描述更优,而且全原子模型计算成本较高,尤其对于长链正构烷烃.然而,到目前为止,详细比较不同常用联合原子势函数对正构烷烃物理性质预测精度的研究比较缺乏.
本文选取了内燃机燃料中典型组分正庚烷和正十二烷为研究对象,比较了不同势函数对正构烷烃物理性质(包括饱和及输运物性)的影响,并提出了一种新的修正力场并对其进行了烷烃饱和两相密度、饱和蒸气压和自扩散系数等热物理性质的验证.另外,将改进力场应用于双组分燃料(正庚烷和正十二烷)热物性的研究中,主要探究了不同混合比例条件下通过将每种组分饱和物性按摩尔比例加权平均的方法来估算混合物饱和物性的准确度.
1 模拟方法
1.1 势函数模型
分子动力学模拟中,势函数或者力场模型是作为粒子间作用力的近似描述,力场的选取对于整个模拟的过程可谓重中之重,因此在进行模拟前需要选择准确合适的力场.每种物质有各自不同适合于本身的力场参数,同一种物质也有适合于表述各种性质而对应的不同的力场参数.为了节约计算成本,本文采用了常用的4种适用于碳氢化合物的联合原子力场模型OPLS-UA、SKS、TraPPE-UA以及NERD.此外,基于SKS非键势能、键伸缩势能以及键角弯曲势能和另一种五参数约束的二面角扭曲势能[10],本文提出一种新的修正力场(命名为SKS-New).
联合原子力场的非键势能可以用截断的Lennard-jones12-6势来描述:

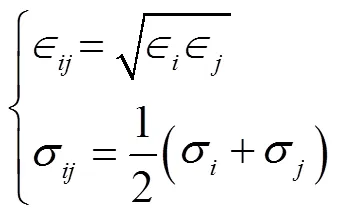
在最初的SKS和TraPPE-UA的力场模型中,将所有键视为固定长度的刚性键,然而LAMMPS平台不提供正构烷烃等大分子的键长约束,因此本研究中统一使用谐波势来代替刚性键拉伸描述键伸缩势能:
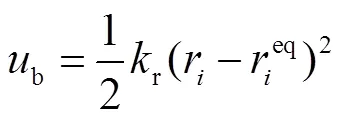
同样用一种谐波势来描述键角弯曲势能:

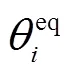
用OPLS二面角扭曲势[4]描述前4种力场的二面角扭曲势能:

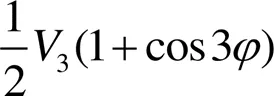
改进力场的二面角扭曲势能则由余弦多项式表示的多谐波势[10]来描述:
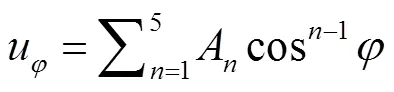
各联合原子模型力场的4项势能参数列于表1.
1.2 热物性统计方法及计算公式


表1 不同力场的势参数
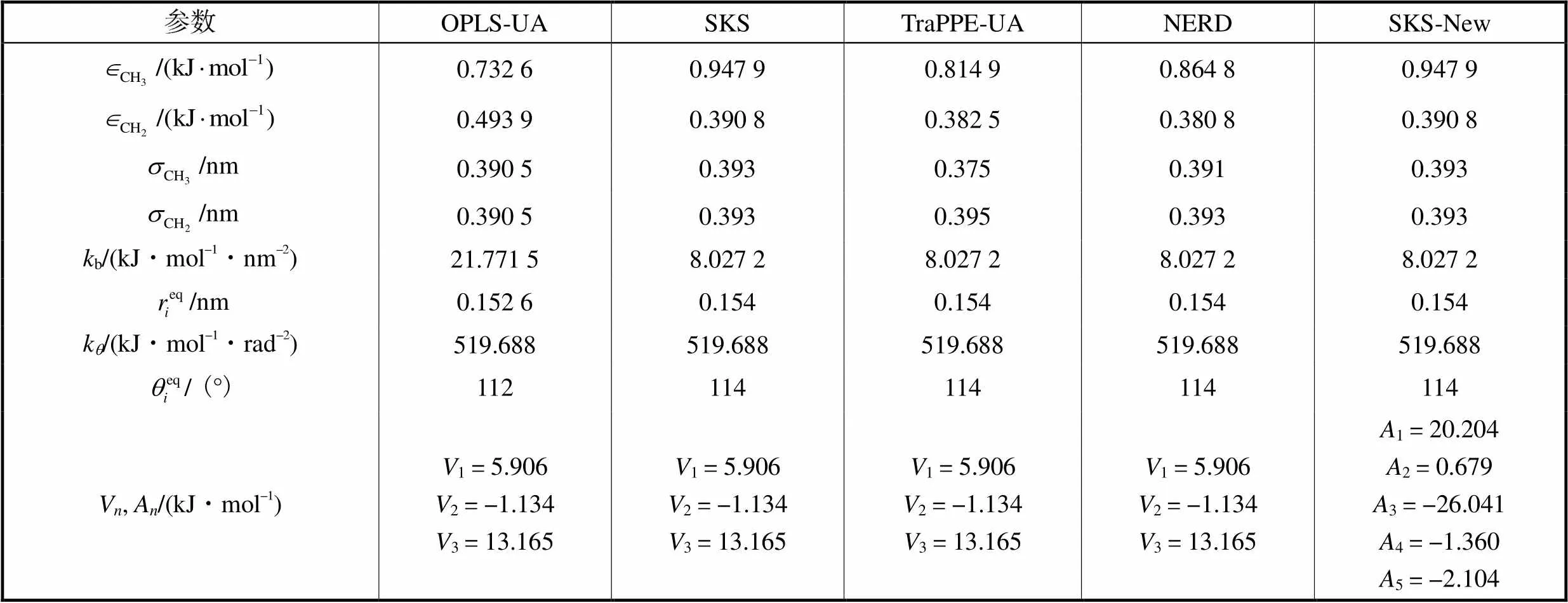
Tab.1 Potential parameters for different force fields

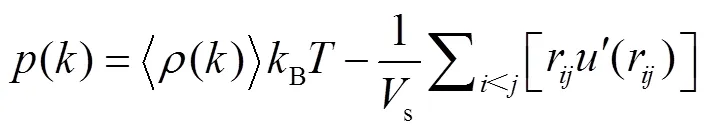
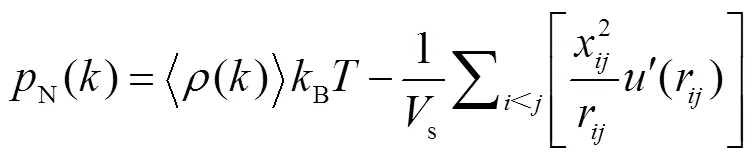
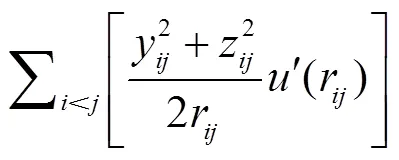
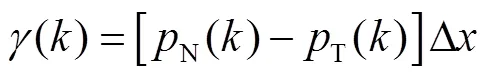


1.3 模拟设置
本文比较了5种力场对从低温到接近临界温度(正庚烷临界温度为540K、正十二烷临界温度为658K)下气液平衡模拟纯组分正构烷烃饱和物性和纯液相烷烃的自扩散特性的影响以及研究了不同摩尔比(正庚烷:正十二烷)的双组分烷烃在正庚烷临界温度以下的饱和物性.系统的初始构型如图1所示,初始构型气液气构型用于饱和物性的计算;纯液相构型用于自扩散系数的计算.因考虑实际计算效率和对结果的影响,不同的模拟过程选择不同的适合的分子数以及模拟盒尺寸,详细模拟细节在表2中总结.
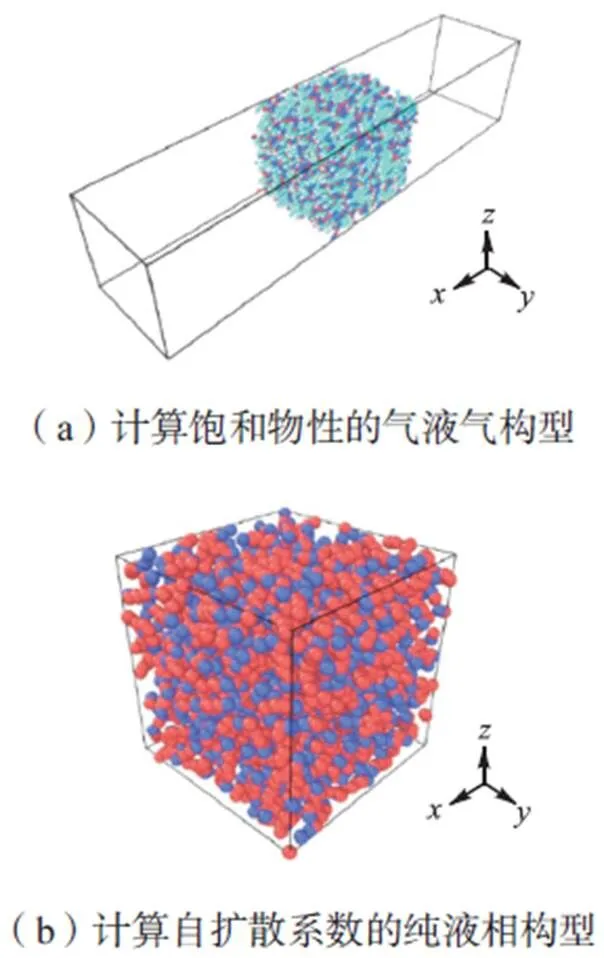
图1 初始构型
表2 不同模拟过程的初始设置

Tab.2 Initialsettings for different simulation processes
气液气构型中模拟盒子为长方体,初始时刻液相分子位于盒子中心,两端为气相区.为消除模拟体系的规模限制带来的边界效应,在模拟盒3个方向上均采用周期性边界条件,整个模拟过程均使用正则(NVT)系综对系统温度进行恒定.采用velocity-verlet算法对原子运动方程进行积分,原子初始速度按高斯分布随机分配.取时间步长为2×10-6ns,模拟总时长为3ns,其中前2ns为弛豫时间,后1ns为统计时间.当整个系统的温度、压力、总能和势能4个指标仅在固定值附近波动时,则认定达到平衡状态.纯液相模型为立方模拟盒,选择合适的模拟盒尺寸,使初始构型的液体密度在不同温度下接近真实值,本次模拟中初始密度取自NIST数据库[13].先用NPT系综运行1ns使整个系统压力稳定在常压附近,然后再使用NVT系综运行2ns,同样取最后1ns作为统计时间.
2 结果与讨论
2.1 纯正庚烷的气液平衡MD模拟
气相与液相的相交处为气液界面,体系平衡状态下即可获得稳定界面.MD模拟很好地再现了气液两相平衡特征,各力场下气液相密度分布曲线如图2所示,在整个体系达到平衡之后,根据这些分布,可以识别出密度较高的液相区域、密度较低的气相区域和气液界面.5种联合原子力场在低于473K以下时都有较好的模拟表现,能够完整地模拟正庚烷从293~473K的饱和过程,有明显的密度梯度存在,温度的升高导致液相的密度下降,而导致气相的密度增加;而在高于473K时,因接近于临界温度(540K),密度梯度缩小而界面区慢慢消失,5种力场中只有OPLS-UA和改进力场SKS-New能完整模拟所有给定温度下的气液平衡过程,在高于473K时也能有明显的气液密度梯度,SKS力场在533K时密度梯度消失,而TraPPE-UA和NERD力场在513K和533K时模拟效果较差,已全部变为气相.
2.2 对纯组分正庚烷和正十二烷的热物性验证
2.2.1 饱和物性
由于界面轮廓随时间波动,气液界面的位置在不同的模拟中有所不同.尽管有些波动,气液界面的统计位置依然稳定.平均统计了5种力场、两种纯组分在各温度下两边气相区密度和中间液相区密度,来与NIST数据库[13]的两相密度进行对比.所得气液共存曲线如图3所示.正庚烷气液共存曲线5种力场的模拟结果总体上大都与NIST值相符,其中TraPPE-UA和NERD力场在413K以下的低温区气液两相区吻合较好,在高于413K时误差增大,气相区密度与NIST值相比偏大而液相区密度偏小,由于二者在513K以上之后就全部变为气相,因此无513K以上的气液共存密度,SKS力场在533K时亦是如此.OPLS-UA力场对所有温度下的液相密度模拟结果都略微偏大,而SKS及提出的改进力场的模拟结果与NIST值最为吻合,但由于新力场在533K时还能保持良好的气液两相平衡,说明改进力场对临界温度的预测效果最好.因为选取精确的势函数是表征模拟准确性的前提,力场描述的粒子间吸引力越小,在给定温度下从中心液核弛豫到气相区的分子越多,使得液相区密度偏小而气相区密度偏大,从模拟结果来看,OPLS-UA力场表述的正构烷烃分子吸引力偏大,TraPPE-UA和NERD力场则偏小,而SKS和改进力场相对而言最为准确.
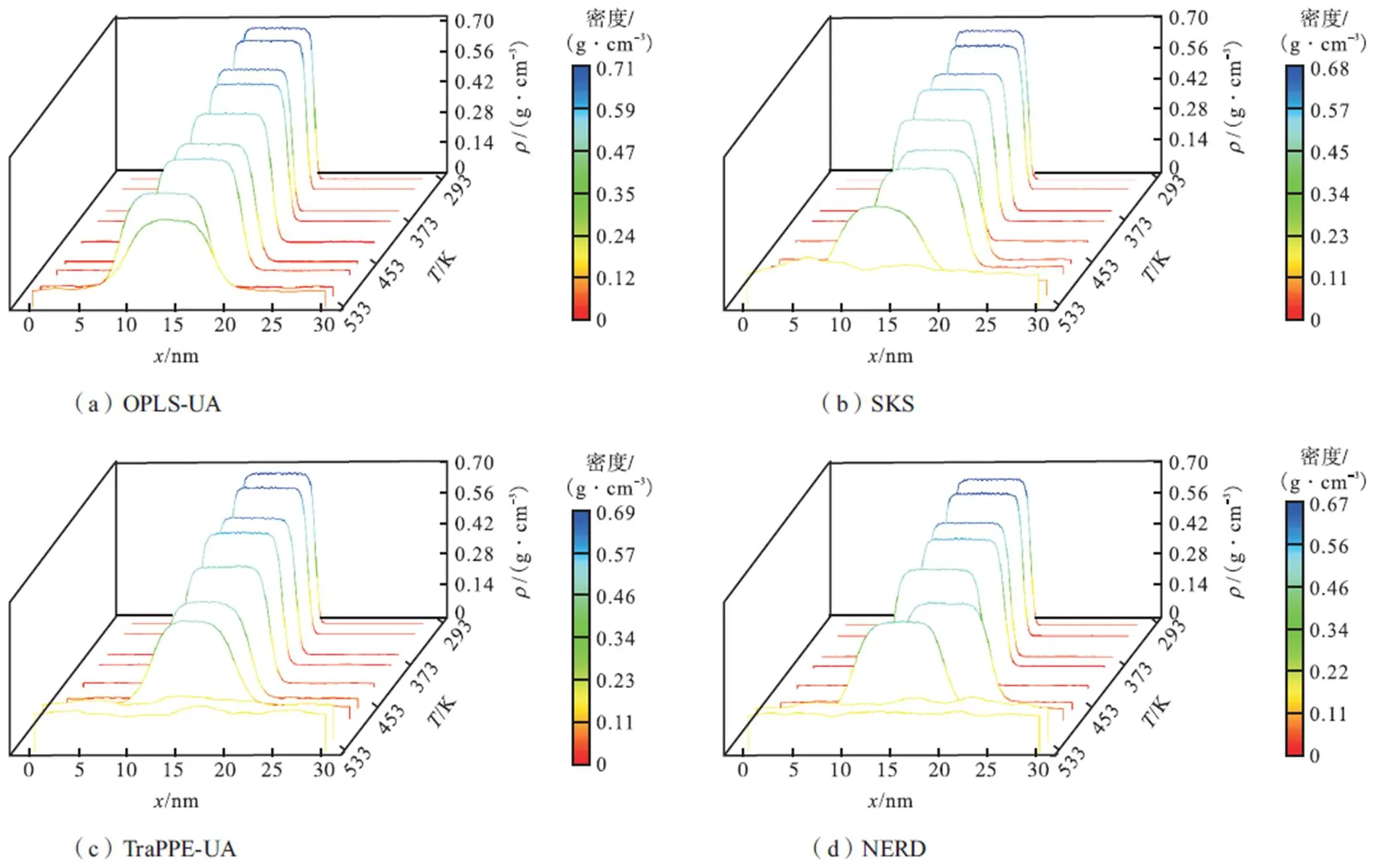

图3 不同力场下的气液共存曲线
不同力场下正十二烷从450~650K的模拟结果与正庚烷大致趋势相同,同样是SKS和改进力场的模拟结果与NIST值较为符合,同样SKS、TraPPE-UA和NERD这3种力场在温度升高时气相密度会偏大、液相密度会偏小,而OPLS-UA力场则相反.从气液共存密度的模拟结果来看,改进力场SKS-New对正十二烷的描述优于其他4种力场,改进力场在每个温度下的模拟结果能相对比较准确地模拟出气液相密度,且在650K即接近临界温度(658K)时也能有明显气液界面,在5种力场中甚至比SKS力场对高温度下的描述更为准确,预测临界点的效果更好.这可能是由于改进力场势能参数所用的二面角扭转势能参数比原SKS力场更为准确,同时在链更长的烷烃里所包含的二面角更多,因此对二面角扭曲势能更为准确的描述会使得模拟结果更为精确.
燃料的饱和蒸气压对于发动机内混合气形成以及后续燃烧过程有着重要影响,因此为验证此改进力场参数是否能准确表述烷烃其他物性,用气液气模型计算了烷烃的不同温度下的饱和蒸气压.将气液平衡状态下两端饱和气相区的压力统计平均值即可得到饱和蒸气压,MD模拟正庚烷和正十二烷的计算结果与实验值[14-15]和NIST数据库[13]的对比如图4所示.两种纯组分不同力场下的饱和蒸气压都随温度升高而增大,模拟结果与前面两相密度的效果大致相同,5种力场中还是SKS和改进力场SKS-New的模拟结果相对较为准确,其他3种力场与实验值或数据库数据偏差较大,尤其在对正十二烷的模拟中更加明显.SKS和改进力场模拟的饱和蒸气压都随温度升高而误差增大,而对于正十二烷改进力场的饱和蒸气压在高温区也能较好地吻合且比SKS力场表现更好,这也印证了上面对于改进力场势能参数更适用于表述中长链烷烃的推断.

图4 不同力场下的饱和蒸气压
2.2.2 纯液相的自扩散系数
除了对气液平衡体系下正构烷烃进行不同力场的MD模拟验证外,还通过建立纯组分纯液相的立方初始构型来模拟计算液相的自扩散系数,用不同力场模拟的正庚烷和正十二烷结果与实验值[16-17]对比分别如图5(a)和(b)所示.自扩散系数的总体趋势随着温度升高而增大,与前面气液模型计算的两相密度和饱和蒸气压效果不同,在5种力场中最为相符的是OPLS-UA力场,其次为SKS-New改进力场和SKS力场,而另外两种力场还是与实验值相比偏大.结合上一小节的研究来看,这可能是因为OPLS-UA力场描述的粒子间吸引力在5种力场中最大,因此模拟过程中分子在自由运动中的均方位移最小,通过均方位移计算的自扩散系数也就最小,而其他力场对应的描述的粒子间吸引力越小,则计算的自扩散系数越大.

图5 不同力场下的液相自扩散系数
2.3 正庚烷/正十二烷二元组分的饱和物性

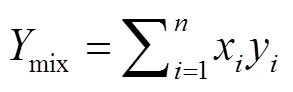
式中:为纯组分的摩尔分数,为纯组分物性的模拟值.结果如图6所示,混合物物性并不是单组分物性的简单加权平均,由于正十二烷分子对正庚烷分子的吸附作用,本该在接近临界温度时全部变为气相的正庚烷分子比预计更少地从液核弛豫到气相区,因此液相密度的加权值始终比实际模拟值偏小,相应地,气相密度和饱和蒸气压始终偏大,且总体趋势似乎是随着温度增大而偏差越多.


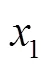

图7 相对误差随温度、混合比的变化关系
3 结 论
采用MD模拟比较了不同势函数对不同温度下纯组分正庚烷和正十二烷燃料物理性质的影响,并提出了一种新的修正力场并对其进行了烷烃饱和两相密度、饱和蒸气压和自扩散系数等热物性的验证.还将改进力场应用于二元组分烷烃的模拟中,主要研究了不同摩尔比(1∶2、1∶1、2∶1)的双组分烷烃在正庚烷临界温度下的饱和过程.本文的研究结果如下.
(1)建立了单组分烷烃气液平衡的MD模型.比较了不同势函数对于预测两种正构烷烃两相饱和物性的精度差异,分析了温度对于饱和状态下两相密度以及饱和蒸气压的影响.随温度升高,液相密度减小,气相密度与饱和蒸气压增大,5种力场皆能良好地模拟烷烃在近临界温度下的饱和过程.
(2)提出了一种改进的适用于正构烷烃的修正力场,比较了与其他4种常用力场对预测饱和两相密度、饱和蒸气压以及自扩散系数的精度.其中SKS力场对于正庚烷的饱和性质模拟最为准确,但对于临界点的预测不如改进力场SKS-New;改进力场对于正十二烷的饱和性质模拟相较于SKS力场要更优,且对两种正构烷烃的临界温度的预测更准确;OPLS-UA力场对于自扩散系数的模拟最为准确,其次为改进力场.综合来看,改进力场对正构烷烃的粒子间作用力描述最优且更适用于中长链烷烃.
(3)研究了双组分烷烃饱和过程.探究了不同混合比和温度条件下通过将纯组分饱和物性按不同摩尔比加权平均的方法估算混合物饱和物性的准确度.在估算饱和物性时,选择温度较低、正庚烷比重较高或者是温度较高、正庚烷比重较低的工况下加权平均更为准确.
[1] Jorgensen W L,Tirado-Rives J. The OPLS[optimized potentials for liquid simulations] potential functions for proteins,energy minimizations for crystals of cyclic peptides and crambin[J].,1988,110(6):1657-1666.
[2] Jorgensen W L,Maxwell D S,Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids[J].,1996,118(45):11225-11236.
[3] Jorgensen W L. Optimized intermolecular potential functions for liquid alcohols [J].,1986,90(7):1276-1284.
[4] Jorgensen W L,Madura J D,Swenson C J. Optimized intermolecular potential functions for liquid hydrocarbons[J].,1984,106(22):6638-6646.
[5] Siepmann J I,Karaborni S,Smit B. Vapor-liquid equilibria of model alkanes[J].,1993,115(14):6454-6455.
[6] Siepmann J I,Karaborni S,Smit B. Simulating the critical behaviour of complex fluids[J].,1993,365:330-332.
[7] Martin M G,Siepmann J I. Transferable potentials for phase equilibria(1):United-atom description of n-alkanes[J].,1998,102(14):2569-2577.
[8] Nath S K,Escobedo F A,de Pablo J J. On the simulation of vapor-liquid equilibria for alkanes[J].,1998,108(23):9905-9911.
[9] 邓 磊. 亚/超临界环境下气液界面特性的分子动力学模拟[D]. 大连:大连理工大学能源与动力学院,2015.
Deng Lei. Molecular Dynamics Simulation of Liquid-Gas Interface in Sub/Supercritical Surroundings[D]. Dalian:School of Energy and Power Engineering,Dalian University of Technology,2015(in Chinese).
[10] Kalyanasundaram V,Spearot D E,Malshe A P. Molecular dynamics simulation of nanoconfinement induced organization of n-decane[J].,2009,25(13):7553-7560.
[11] Mo G,Qiao L. A molecular dynamics investigation of n-alkanes vaporizing into nitrogen:Transition from subcritical to supercritical[J].,2017,176:60-71.
[12] Lee S-H. Pressure analyses at the planar surface of liquid-vapor argon by a test-area molecular dynamics simulation[J].,2012,33(9):3039-3042.
[13] NIST. NIST Chemistry WebBook[EB/OL]. https:// webbook.nist.gov/chemistry/,2022-03-16.
[14] Weber L A. Vapor pressure of heptane from the triple point to the critical point[J].,2000,45(2):173-176.
[15] Morgan D L,Kobayashi R. Direct vapor pressure measurements of ten n-alkanes in the C10-C28 range[J].,1994,97:211-242.
[16] Fishman E. Self-diffusion in liquid n-pentane and n-heptane[J].,1955,59(5):469-472.
[17] Harris K R,Ganbold B,Price W S. Viscous calibration liquids for self-diffusion measurements[J].,2015,60(12):3506-3517.
Molecular Dynamics Simulation of Physical Properties of n-Alkane Based on Different Potential Models
Chen Feilong1,Zhang Yanzhi1,He Zhixia2
(1. School of Energy and Power,Dalian University of Technology,Dalian 116024,China;2. Institute for Energy Research,Jiangsu University,Zhenjiang 212013,China)
In this paper,molecular dynamics simulation was used to compare in detail the effect of four commonly used potential models on the prediction accuracy of the physical properties of n-alkane,and a new modified model was proposed and extensively validated,which mainly includes the thermophysical and transport properties such as saturated two-phase density,saturated vapor pressure and self-diffusion coefficient. After that,the modified potential model was applied to the study of the thermophysical properties of binary-component fuels,and the accuracy of estimating the saturated physical properties of the mixture by the weighted average method of each component at different mixing ratios was explored. It was found that the predictions from the modified model show good agreement with the measurements for n-heptane and n-dodecane. When the temperature is lower and the mole fraction of n-heptane is higher,or when the temperature is higher and the mole fraction ofnheptane is lower,the saturated physical property value obtained by the weighted average is more accurate.
molecular dynamics simulation;physical property;potential model;binary-component
10.11715/rskxjs.R202305023
TK401
A
1006-8740(2023)04-0451-09
2022-03-16.
国家自然科学基金资助项目(52106154);江苏省自然科学基金资助项目(BK20190855).
陈飞龙(1998— ),男,硕士研究生,fl_chen98@163.com.
张延志,男,博士,副教授,zhangyanxzhi@dlut.edu.cn.
(责任编辑:隋韶颖)
猜你喜欢
航空材料学报(2023年6期)2023-12-18 05:23:50
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
纺织标准与质量(2022年3期)2022-08-10 09:11:22
中国音乐学(2022年1期)2022-05-05 06:48:46
云南化工(2021年11期)2022-01-12 06:06:30
岩矿测试(2021年3期)2021-07-06 06:04:00
物理学报(2018年10期)2018-06-14 06:31:32
草业学报(2016年6期)2016-09-01 07:16:49
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:06
合成化学(2015年9期)2016-01-17 08:57:02
