
靶向PD-L1蛋白的计算机辅助药物筛选*
2024-01-06 10:24林开东林晓倩2林绪波
物理学报 2023年24期
林开东 林晓倩2) 林绪波†
1) (北京航空航天大学,医学科学与工程学院/生物与医学工程学院,北京市生物医学工程高精尖创新中心,北京 100191)
2) (北京航空航天大学沈元学院,北京 100191)
1 引言
阻断免疫检查点蛋白与其配体的结合作为肿瘤治疗的方法之一,近年来在临床应用中迅速发展.正常生理状态下,部分负调节因子作为免疫检查点来抑制T细胞的过度激活,确保免疫反应保持自我耐受[1].然而不幸的是,肿瘤细胞可以利用这种机制诱导T细胞衰竭,形成促进肿瘤生长和侵袭的微环境,从而逃避免疫系统的攻击[2-4].为了重新激活并增强T细胞介导的抗肿瘤功能,前人已经设计了一系列疗法来阻断免疫检查点蛋白与其配体的结合[5,6].其中,细胞程序性死亡蛋白I(programmed cell death protein 1,PD-1)是最受关注的免疫检查点之一,其通常表达于活化的T细胞、自然杀伤性细胞、B淋巴细胞和其他免疫细胞的表面.PD-1与其在肿瘤细胞上高度表达的配体PD-L1相互作用后,发生一定的构象变化,介导胞内信号通路从而抑制T细胞的增殖、活化和细胞杀伤性功能[7-12].前人的研究已表明,PD-1或PD-L1的基因敲除或抗体抑制可以增强小鼠免疫系统的抗肿瘤功能,这表明阻断PD-1和PDL1之间的相互作用可能为肿瘤免疫治疗提供一种有效的策略[13,14].
PD-1/PD-L1抗体抑制剂药物的研发目前取得非常显著的进展.2014年,美国食品药品监督管理局批准了第一款PD-1抗体Pembrolizumab用于治疗晚期黑色素瘤之后,一系列PD-1/PD-L1单克隆抗体被应用在了非小细胞肺癌、肝癌和食管胃交界癌等多种肿瘤疾病的临床治疗中[15-17].然而,随着应用的不断深入,抗体半衰期长、慢性免疫毒性、组织渗透有限、存储和运输成本高等难以避免的缺点逐渐暴露出来[18-20].另一方面,抗体的同质化研究过多,造成了较大的资源浪费.小分子抑制剂具备较好的肿瘤组织渗透性、相对稳定的生物安全性和良好的口服利用性等优势,已成为PD-1/PD-L1抑制剂药物的下一个研发热点[18,21-23].
百时美施贵宝(Bristol-Myers Squibb,BMS)于2015年公开了一系列非肽基联苯类小分子抑制剂,对PD-1/PD-L1结合具有强大的阻断抑制活性[24].Holak团队[25]曾报道,BMS化合物通过与PD-L1结合诱导其发生二聚化,以间接的方式抑制PD-1/PD-L1相互作用.基于这一机制,其他的公司和学术团队开发了一系列不同骨架的衍生物,如Incyte公司的INCB086550 (NCT04629339/NCT 03762447)[26]、红日药业的IMMH-010 (NCT0434 3859)[27,28]、再极药业的MAX-10181 (NCT04122 339)、贝达药业的BPI-371153 (NCT05341557)、歌礼药业的ACS61 (NCT05287399)以及和誉生物医药的ABSK043 (NCT04964375)等化合物目前已进入到了临床试验阶段.由于目前PD-L1小分子抑制剂的市场空白,寻找更多样骨架且具备良好用药性质的化合物仍具备重要意义.
数据驱动的计算方法已成为药物开发的重要工具,PD-1/PD-L1小分子抑制剂亦不例外[29].基于结构的方法,如药效团分析、分子对接和分子动力学(molecular dynamics,MD)模拟,在先前的研究中被广泛应用于靶向PD-L1二聚体的小分子抑制剂的虚拟筛选[29-33].本文基于各种机器学习算法和分子描述符或指纹构建了一系列基于配体方法的分类和回归模型,以预测ZINC15[34]中类药物化合物对PD-1/PD-L1相互作用的抑制活性.具有高预测活性的化合物将继续通过药物相似性、药代动力学筛选并以分子对接和MD模拟进行基于结构方法上的验证,以最终获得具备PD-L1抑制潜力的小分子化合物.
2 研究方法
2.1 数据获取及整理
本文用于训练及测试的数据集来源于与PDL1小分子抑制剂相关的37篇研究性论文及16项专利(见补充材料表S1 (online)),为避免因实验技术手段不同而导致化合物对PD-L1抑制活性测定数据水平不一致的情况,本文仅收录了以半抑制浓度(half-maximal inhibitor concentration,IC50)为指标的均相时间分辨荧光(homogeneous timeresolved fluorescence,HTRF)的实验数据.
参考研究性论文及专利中的结构示意图,本文利用ChemDraw获取并检查化合物的简化分子线性输入规范(simplified molecular input line entry system,SMILES)字符串.IC50不高于1 μmol/L的化合物定义为阳性样本(即对PD-L1具备抑制活性),而IC50高于10 μmol/L的化合物定义为阴性样本(即对PD-L1不具备抑制活性),为减小用于分类模型构建的两类样本数量不平衡问题,一项细胞水平的高通量筛选(high throughput screening,HTS)实验记录(PubChem BioAssay AID: 2316)部分数据被引入补充阴性样本.最后,仅具备明确IC50测定值而非测定范围的化合物被收录于回归模型的数据集中,IC50的对数转化值pIC50(即-lgIC50)作为样本标签.
2.2 数据集聚类
为了确保机器学习模型尽可能均匀地获得数据集中不同结构的信息,本文对数据集中的化合物先进行了聚类处理.首先,2130个分类模型阳性样本和1099个回归模型样本分别转化为600和350种仅保留环形结构和连接环形结构的最短路径的Murcko骨架[35].分子骨架以2为最大半径,2048为向量长度转化为圆形扩展指纹(extendedconnectivity finger print,ECFP)[36]后,以平均为链接算法、欧氏距离为计算方式对两类骨架的ECFP进行分层聚类,簇的数量由5—20之间对应的最佳轮廓系数决定,后续模型训练将从簇内分层抽样进行数据集划分(图1).

图1 数据集聚类 (a)分类模型阳性样本骨架的14个簇;(b)回归模型样本骨架的13个簇.环形树状图的颜色代表不同的分子结构骨架簇,颜色越接近,骨架越相似,一旁的化学结构图是每个簇中最具代表性的骨架Fig.1.Dataset clustering: (a) 14 clusters of scaffolds of active compounds in the classification models;(b) 13 clusters of scaffolds of compounds in the regression models.The colors of the circular dendrograms represent different clusters of scaffolds,and the closer the colors are,the more similar the scaffolds are.The chemical structure diagrams on the side are the most representative scaffolds of each cluster.
2.3 分子表征
由ChemDraw得到的化合物SMILES字符串分别转化为三类分子描述符(RDKit,PaDEL 1D&2D,Mordred)和三类分子指纹(ECFP,MACCS,PubChem)以作为化合物的特征向量.开源Java软件PaDEL-Descriptor v2.0用于计算PaDEL 1D&2D描述符[37]、MACCS分子指纹[38]和Pub Chem分子指纹[37].RDKit描述符和ECFP分子指纹[36]均由化学信息处理程序包RDKit转化,其中ECFP分子指纹为2048维向量,指示最大以两个原子为半径的结构碎片存在与否.此外,描述符计算软件Mordred[39]被用于最后一类特征向量的转化.
2.4 机器学习
分别使用五种机器学习算法构建PD-L1小分子抑制剂分类模型和抑制活性IC50预测回归模型,其中逻辑回归(logistic regression,LR)、邻近算法(K-nearest neighbor,KNN)、支持向量机(support vector machine,SVM)、随机森林(random forest,RF)和多层感知机(multilayer perceptron,MLP)用于分类任务模型构建,SVM、岭回归(ridge regression)、高斯过程回归(Gaussian process regression,GPR)、RF和MLP用于回归任务模型构建.两类模型数据集的80%为训练数据集,另外20%为测试数据集.所有分子描述符及分子指纹的特征值删除空缺之后,基于训练集进行min-max标准化处理.各类算法的最佳超参数由基于Scikitlearn网格搜索方法的五重交叉验证(5-fold cross validation)确定,此过程中,分类模型以马修斯相关系数(matthews correlation coefficient,MCC),回归模型以预测值的平均绝对误差(mean absolute error,MAE)为超参数选择标准.
2.5 模型评价标准
分类模型性能由灵敏度(sensitivity,SE)、特异度(specificity,SP)、准确度(accuracy,ACC)、马修斯相关系数(Matthews correlation coefficient,MCC)进行泛化评估,其计算公式如下:
其中,TP(true positive)为分类模型判定为具备抑制活性且实验结果亦为具备抑制活性的化合物样本数,FP(false positive)为分类模型判定为具备抑制活性但实验结果为不具备抑制活性的化合物样本数,TN(true negative)为分类模型判定为不具备抑制活性且实验结果亦为不具备抑制活性的化合物样本数,FN(false negative)为分类模型判定为不具备抑制活性但实验结果为具备抑制活性的化合物样本数.
回归模型性能由平均绝对误差(mean absolute error,MAE)、均方根误差(root mean-square error,RMSE)和决定系数(correlation coefficient,R2)进行泛化评估,其计算公式如下:
其中,n为回归模型待评估数据集样本数,Ytrue为化合物pIC50实验测定值,Ypred为回归模型对化合物pIC50的预测估计值.
2.6 药物相似性及ADMET检验
本文选取来自ZINC15数据库[34]的7400926个可商业购买(截至2023年2月24日)、电中性且收录三维结构信息的类药小分子作为候选化合物筛选池,尽管这些分子已通过ZINC15的药物相似性检验,但由于本文所筛选的化合物针对蛋白质相互作用,传统的药物相似性指标(quantitative estimate of drug-likeness,QED)已经不再适用[40].Kosugi和Ohue[41]提出了一种更加适合于针对蛋白质相互作用抑制剂筛选的药物相似性指标(quantitative estimate index for compounds targeting protein-protein interactions,QEPPI),化合物的QEPPI分数为一个介于0—1之间的值,数值越大代表化合物的蛋白质相互作用抑制剂药物相似性越高,本文以0.7为阈值筛选高类药性化合物.
ADMET代表化合物的吸收(absorption)、分配(distribution)、代谢(metabolism)、排泄(excretion)和毒性(toxicity)等重要用药性质,在早期药物筛选中十分重要,本文采用ADMETlab 2.0对化合物进行ADMET筛选[42].
2.7 分子对接
基于结构的活性预测方法需要蛋白质靶点的晶体结构信息,本文选取的PD-L1二聚体结构来源于蛋白质数据库PDB(ID: 7DY7).本文选择PD-L1二聚体中的结合界面区域为对接结合口袋,基于如下理由: 1)前期的研究表明PD-L1二聚体中的结合界面区域是很有潜力的药物结合位点[43,44];2)分子对接结果显示小分子在该区域的结合亲和力最强.
分子对接利用AutoDock Vina进行[45],Auto-DockTools将PD-L1二聚体转化为pdbqt格式,对接网格盒子尺寸为40 Å×30 Å×40 Å,中心坐标为(144.799 Å,-9.364 Å,16.163 Å).每个化合物各随机生成50种对接姿态和位置,根据对接得分进行排名,最佳得分前十位小分子对应的结合构象将用于分子动力学模拟的初始构象.此外,为验证通过上述流程得到的候选化合物作用机制是否与前人的研究结果相近,本文利用LigPlot+研究小分子与PD-L1二聚体的相互作用[46].
2.8 分子动力学模拟
本文所有的分子动力学模拟采用CHARMM 36全原子力场[47,48],化合物配体的力场参数通过CGenFF程序获取[47-50].十个高对接分数的候选化合物和两个对照化合物(BMS202以及INCB086550)与PD-L1二聚体复合系统搭建于10nm×10nm×10nm盒子内,每个盒子填充水分子并以NaCl中和体系电荷数,整个搭建过程由GROMACS[51]工具gmx solvate和gmx genion实现.所有的分子动力学模拟工作均使用GROMACS 2019.6程序包运行,模拟步长为2.0 fs,采用等温等压(NPT)系综,模拟时长为100 ns.模拟盒在x轴、y轴和z轴3个方向上均设定了周期性边界条件.采用半各向同性的Parrinello-Rahman[52]方法将系统压强维持在1 bar (1 bar=105Pa),压缩系数设定为4.5×10-5,弛豫时间为5 ps.此外,系统采用Nose-Hoover[53,54]控温方法将配体-蛋白质复合物和溶剂进行耦合,温度维持在310 K,弛豫时间为1 ps.长距离静电相互作用使用Particle-Mesh Ewald (PME)[55]方法计算,短距离静电相互作用的截止距离设置为1.2 nm,LINCS (LINear constraint solver)[56]算法用于约束含H原子的键长.
2.9 MM/PBSA结合自由能计算
本文以gmx_MMPBSA[57]为工具利用分子力学泊松-玻尔兹曼表面积法(molecular mechanics Poisson-Boltzmann surface area,MM/PBSA)来计算各个体系最后10 ns轨迹的平均结合自由能(ΔG),其计算公式如下:
其中Gcomplex为蛋白质-配体复合物的自由能,Gprotein和Gligand分别为蛋白质和配体各自的自由能.各组分的自由能计算公式如下:
其中EMM为气相结合能量,由范德瓦耳斯项Evdw和静电项Eele构成;Gsol为溶剂化自由能,由极性EPB和非极性ESA贡献构成;TΔS为熵变,因其计算成本较高且未必能够提高自由能的精度,本文暂不对其进行计算.
3 研究结果
3.1 分类模型性能
30种分类模型分别基于LR,KNN,SVM,RF和MLP五种算法以及RDKit,PaDEL 1D&2D,Mordred分子描述符和ECFP,PubChem,MACCS分子指纹6种分子表征方式所建立.图2为各个模型经五折验证并设置最佳超参数后在测试集上的性能表现,其中以ECFP为输入的KNN模型表现出最佳性能,SE(阳性样本预测正确率)=0.9937,SP(阴性样本预测正确率)=0.9781,ACC(全部样本预测正确率)=0.9859,MCC(综合评价两类样本预测性能指标)=0.9720.考虑到分子描述符或分子指纹的计算较为耗时,同时获取740万余个类药小分子的全部六种输入向量的计算量更大,因此,仅采用以ECFP为输入用于后续的分类筛选.另一方面,相对于LR和MLP两种模型,KNN,SVM和RF模型对于可能具备噪声数据的任务具有较好的鲁棒性,因此,选择KNN,SVM和RF模型作为分类筛选的模型;当有两种或以上的模型支持小分子为活性时,则认定该化合物对PD-L1具备抑制活性.

图2 分类模型性能表现 (a)灵敏度;(b)特异度;(c)准确度;(d)马修斯相关系数Fig.2.Performance of binary models for classification tasks: (a) SE;(b) SP;(c) ACC;(d) MCC.
3.2 分类模型解释
为了进一步探索输入向量的特征与分类之间的相关性,本文用活性和非活性化合物之间的RDKit,PaDEL和Mordred三种描述符来分析基于特征重要性而构建的RF模型中权重最高的5个特征的分布差异(见补充材料图S1 (online)).尽管部分特征在两类样本间的分布差异是十分显著的,但由于分子描述符的特征信息难以解释,特征值根据理化性质及矩阵运算得出,其大小本身不具备具象含义,我们的认知只能停留在较浅薄的层面.
与描述符不同的是,分子指纹ECFP的位点指示局部亚结构的存在或不存在,这可能揭示分子结构与对PD-1/PD-L1的抑制活性之间的关系.分别统计了前十位活性化合物中比例显著高于非活性化合物的子结构和前十位活性化合物中比例显著低于非活性化合物的子结构(图3).带有黄色芳香性原子的环状结构在活性化合物中的计数远多于其在非活性化合物中的计数,这意味着芳香性结构片段可能对化合物的PD-L1抑制活性有正向贡献,相反,在非活性化合物中常出现的带灰色原子的脂肪链片段则对活性没有正向贡献.

图3 分子结构片段在两类样本间的计数差异 (a)活性化合物计数占优的结构片段;(b)非活性化合物计数占优的结构片段.结构片段的中心原子以蓝色突出显示;芳香性原子被着色为黄色,而脂肪烃链原子则被着色为灰色Fig.3.Count difference of fragments of structures between active and inactive compounds: (a) Fragments in active compounds with a proportion higher than inactive compounds;(b) fragments in active compounds with a proportion lower than inactive compounds.The center atoms of substructure fragments are highlighted in blue;aromaticity atoms are colored in yellow and aliphatic hydrocarbon atoms are colored in gray.
值得注意的是,ECFP985和ECFP1161片段清楚地表示了化合物的联苯特性,这也是BMS化合物的核心结构特征之一[24,58].此外,苯甲基(ECFP253)、吡啶(ECFP1453)和与苯环相连的醚键(ECFP1971)通常存在于多数表现出PD-L1抑制活性的小分子结构中.前人的分子对接分析表明,苯甲基能够与PD-L1的Ala121和Met115产生强烈的疏水相互作用[59].Lu等[60]发现PD-L1-BMS202复合物中,吡啶环的氮原子周围有很大的空间,因此他们在该位点附加了一系列的取代基,以提高配体的结合亲和力.由于该片段的重要性,许多其他研究团队也将工作重点放在吡啶的修饰上[59,61,62].除了环状结构,连接环状结构的路径可能也是值得关注的组成部分,其中,以醚键连接六元环或五元环的活性化合物约占50%[43,63-65].
3.3 回归模型性能
30种回归模型分别基于SVM,Ridge Regression,GPR,RF和MLP五种算法以及RDKit,PaDEL 1D&2D,Mordred分子描述符和ECFP,PubChem,MACCS分子指纹6种分子表征方式所建立.图4为各个模型经五折验证后、最佳超参数设置下在测试集上的性能表现,其中以ECFP为输入的SVM模型以MAE=0.4503,RMSE=0.6375,GPR模型以MAE=0.4557,RMSE=0.6375的性能表现显著优于其他模型.图5展示了两种模型在训练集及测试集所有样本点的预测结果及偏差,绝大部分的样本点预测偏差在±1范围内,以ECFP为输入的SVM模型在测试上的决定系数R2=0.782,以ECFP为输入的GPR模型在测试上的决定系数R2=0.787,两类模型预测值的平均值将作为候选化合物的PD-L1抑制活性预测值.

图4 回归模型性能表现 (a)平均绝对误差;(b)均方根误差Fig.4.Performance of continuous models for regression tasks: (a) MAE;(b) RMSE.

图5 两种最佳性能回归模型预测结果 (a),(c)样本预测值与标签值分布;(b),(d)样本预测偏差Fig.5.Prediction results of two best-performing continuous models: (a),(c) Distribution of predicted values and label values;(b),(d) prediction residuals.
3.4 虚拟筛选
ZINC15数据库中电中性且收录三维结构信息的类药物小分子构成本文的化合物筛选池(7400926个小分子).整个虚拟筛选过程包含4个环节: 1)同时使用以ECFP为输入的KNN,SVM和RF等3种分类模型,其中至少有2种判定结果为对PD-L1蛋白具有抑制活性,则认定该分子具有抑制活性.2)同时使用以ECFP为输入的SVM和GPR回归模型预测pIC50值,两模型pIC50的平均值小于7 (IC50 > 100 nmol/L)的化合物将被剔除.3)计算小分子化合物的QEPPI分数(见补充材料表S2 (online)),并保留QEPPI得分超过0.7的候选化合物.尽管初始筛选池的化合物在ZINC15中根据Lipinski五原则[66]被定义为类药化合物,但其中部分化合物因分子量过小可能难以充分结合到PD-L1二聚体的相互作用界面,因此,重新评估他们的药物相似性仍具有重要意义.4)使用ADMETlab 2.0[42]计算小分子的AD MET性质,以进一步筛选具有应用潜能的小分子(见补充材料表S3 (online)).最终,通过整个虚拟筛选流程,42个候选化合物被认为对PD-L1具有较强的抑制活性并具备良好的药用性能(图6).
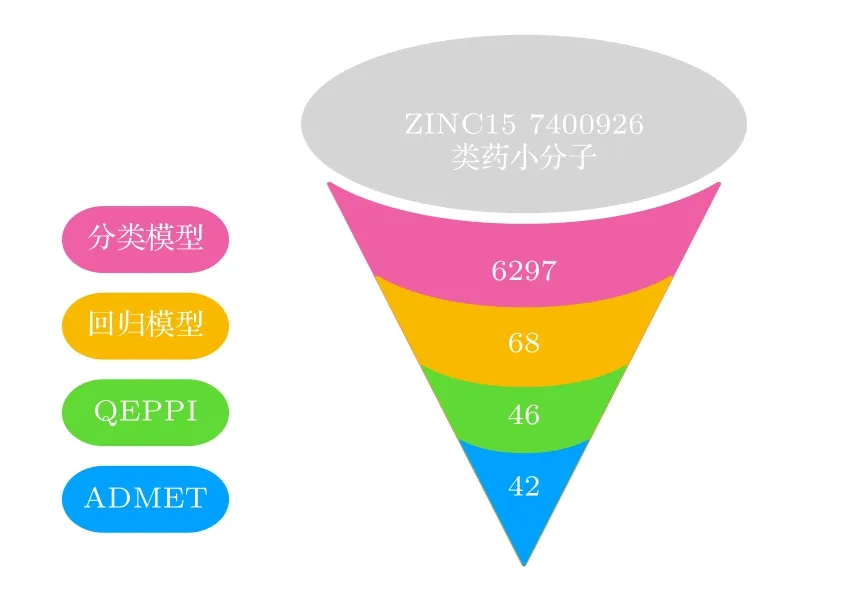
图6 虚拟筛选流程Fig.6.Workflow of virtual screening.
3.5 分子对接
为了验证筛选结果并通过基于结构的方法进一步识别有抑制潜力的化合物,利用AutoDock Vina将42个候选化合物以及BMS202和INCB-086550对接到PD-L1二聚体的IgV结构域(PDB ID:7DY7).具有最低对接评分的10种化合物被认为有潜力的PD-L1抑制剂并继续用于之后的分析(表1),其中值得注意的是,10种候选化合物对接分数均低于临床II期试验化合物INCB086550的对接分数,因此可以认为先前构建的分类和回归模型是筛选PD-L1小分子抑制剂的有效工具.分析12种化合物与PD-L1二聚体的相互作用可知(见补充材料图S2 (online)),TYR56(A),TYR56(B),MET115(A),MET115(B),ALA121(A)和TYR123(A)(括弧中A或B分别表示PD-L1二聚体中的单体A或B)等氨基酸残基与所有配体均发生了较强的疏水相互作用,这意味着这些小分子的结合位置和结合模式可能是近似的.此外,除了ZINC000021874692和ZINC000021874694这一对化合物为旋光异构体,其余化合物的结构差异较大,相似度较低,意味着其可为未来的湿实验筛选提供较高的容错空间(见补充材料图S3 (online)).
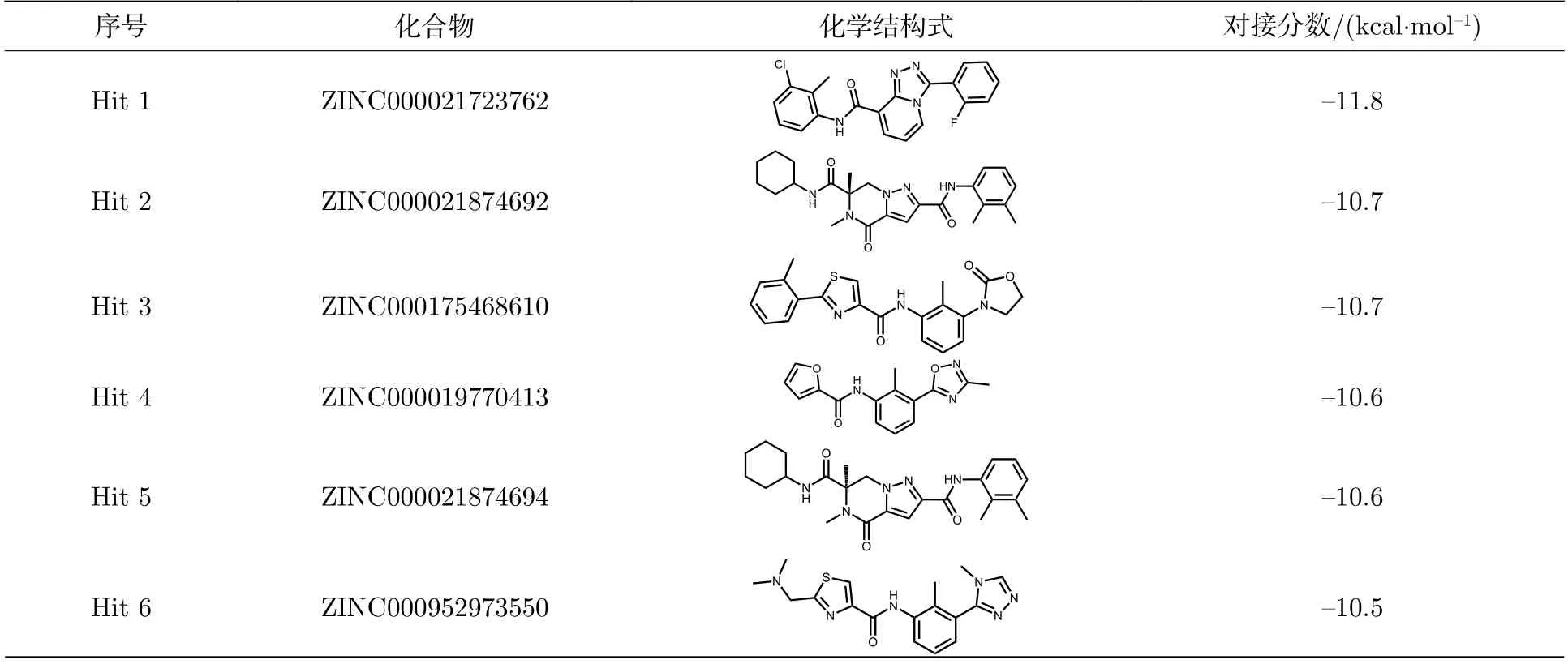
表1 最低对接评分的10种候选化合物及BMS202和INCB086550的对接结果Table 1.Docking results for the top 10 hits with BMS202 and INCB086550.
3.6 分子动力学模拟
为验证配体与PD-L1二聚体结合的稳定性,计算了12种配体-蛋白质复合物100 ns轨迹中蛋白质骨架和配体小分子构象基于蛋白质骨架叠合的均方根偏差(root mean square deviation,RMSD)(图7).所有体系的PD-L1二聚体骨架和配体的RMSD在50 ns后均稳定在0.8 nm以下,模拟轨迹达到平衡,蛋白质与配体结合稳定.值得注意的是,以INCB086550为例的分子量较大的小分子与以ZINC000019770413为例的分子量较小的小分子相比,配体构象RMSD波动较大.由图8的蛋白质-配体结合模式可知,起到关键作用的结构片段集中在配体的一端,而另一端暴露于结合口袋之外.若配体的分子量较大,分子骨架较长,其在结合区域外的部分更多,这部分运动更为自由,这可能是部分小分子构象RMSD波动相对较大的原因.

图7 RMSD (a) PD-L1二聚体骨架;(b)配体Fig.7.RMSD: (a) Backbone of PD-L1 dimer;(b) ligand.

图8 配体与PD-L1结合模式和关键残基 (a) ZINC000021 723762;(b) ZINC000021 874692;(c) ZINC000175 468610;(d) ZINC 000019 770413;(e) ZINC000021 874694;(f) ZINC000952 973550;(g) ZINC000064 987401;(h) ZINC000003 908573;(i) ZINC 000020 538424;(j) ZINC000004 063088;(k) BMS202;(l) INCB086550Fig.8.Ligand binding mode with PD-L1 and key residues: (a) ZINC000021 723762;(b) ZINC000021 874692;(c) ZINC000175 468610;(d) ZINC000019 770413;(e) ZINC000021 874694;(f) ZINC000952 973550;(g) ZINC000064 987401;(h) ZINC000003 908573;(i) ZINC000020 538424;(j) ZINC000004 063088;(k) BMS202;(l) NCB086550.
3.7 MM/PBSA结合自由能计算
分子动力学模拟的最后10 ns轨迹被用于MM/PBSA计算以评估配体小分子与PD-L1二聚体间的结合作用强度.由表2可知,大部分候选化合物的结合自由能均与对照组的结合自由能处于同一水平,而ZINC000021723762,ZINC0000218 74694和ZINC000004063088甚至显著优于对照组水平,范德瓦耳斯相互作用为驱动这三类化合物区别于其他化合物的主要因素.
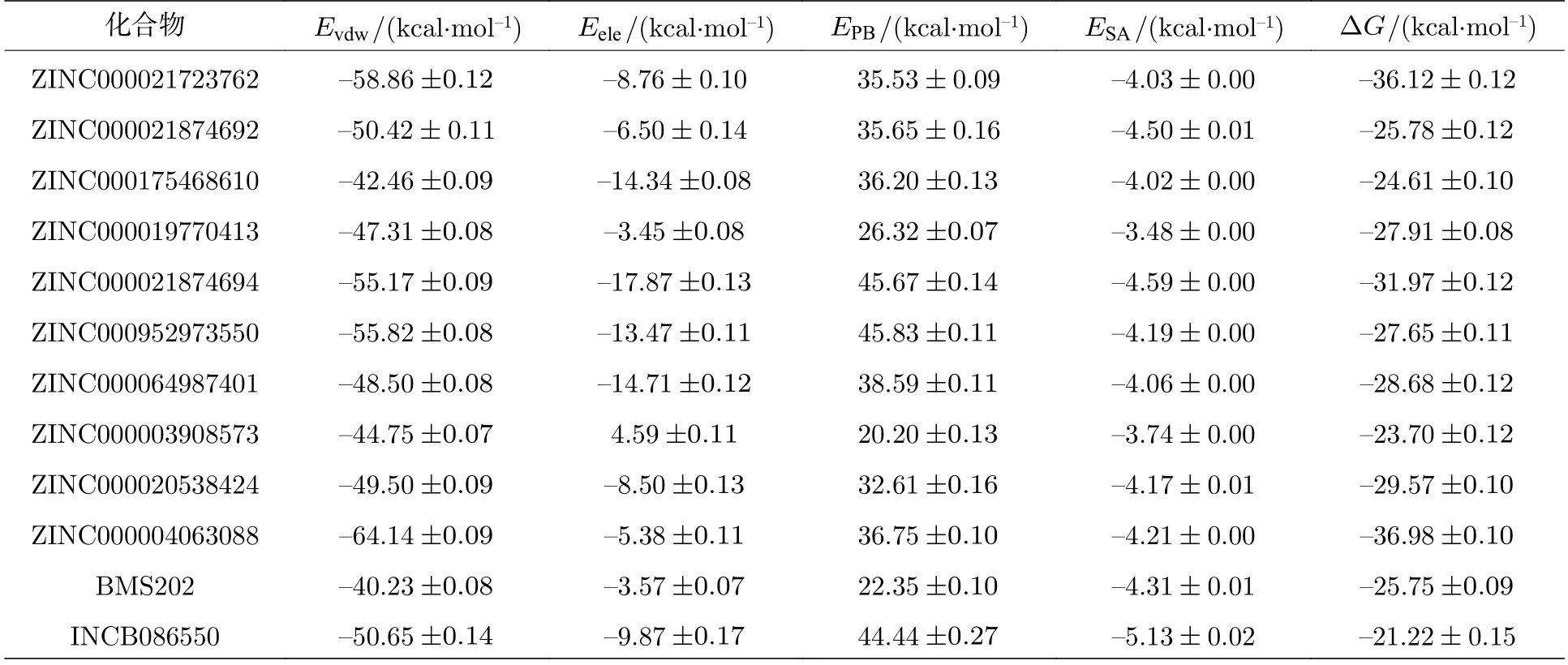
表2 结合自由能计算结果Table 2.Results of MMPBSA.
将结合自由能分解至每个氨基酸残基与配体分子的相互作用上,提取了每个体系中对结合强度贡献度最大的5个关键残基.如图8所示,TYR56(B),TYR123(A),MET115(B),ALA121(A)在绝大部分体系中都起到关键作用,这意味着12种配体结合于PD-L1二聚体相互作用交界面的模式是近似的.前人研究揭示出,MET115(B)和ALA 121(A)与化合物中的芳香环能够发生强烈疏水相互作用,而TYR56(B)能够与化合物的苯环形成π-π堆积以增强结合稳定性[32,33,59,67],这些作用在结合自由能贡献中均得到了体现.
4 结论
通过从相关研究文献及专利收集PD-L1小分子抑制活性数据集,并对数据集内化合物以结构相似性进行分层抽样,本文构建了各30种基于不同分子表征方法和算法的机器学习活性判定分类模型和活性强度回归预测模型,其中性能最佳分类模型分类正确率可达98%以上,回归模型预测平均绝对误差在0.5以下,具备良好的应用价值.将以上模型并结合药用性质筛选工具构成的虚拟筛选方法应用于ZINC15大型小分子数据库,获得了42种有潜力的PD-L1小分子抑制剂,其中的10种通过分子对接、分子动力学模拟和结合自由能估计验证发现,候选化合物的作用机制与对照化合物相近,部分化合物的结合强度甚至优于对照化合物,这意味着本文前期建立的机器学习模型是可以帮助加速PD-L1小分子抑制剂虚拟筛选的有效工具.
然而,完整的药物研发并不是一个简单的工程,本文仅使用计算方法帮助加速PD-L1小分子抑制剂的研发,在完整的研发产业链中处于较上游的位置.为尽可能使得本文的筛选结果得到更进一步的有效认证,后续分子水平、细胞水平以及动物模型水平的湿实验验证仍是必不可缺的,计算机技术目前仅能扮演锦上添花的角色.相信随着生物计算领域与生物技术领域的合作加深,药物研发将能够向一个尽可能高效、低成本的方向发展.
猜你喜欢
上海农业学报(2017年3期)2017-04-10
材料科学与工程学报(2016年4期)2017-01-15
中国卫生标准管理(2015年16期)2016-01-20
合成化学(2015年4期)2016-01-17
中国当代医药(2015年16期)2015-03-01
现代检验医学杂志(2015年2期)2015-02-06
现代检验医学杂志(2015年1期)2015-02-06
中国药理学通报(2014年2期)2014-05-09
无机化学学报(2014年6期)2014-02-28
无机化学学报(2014年5期)2014-02-28
