
生命起源前手性对映体富集与放大的数学规律与实验
2024-01-05 02:45朱华结李志伟贾云静朱玉俊CharlesPittmanJr
河北科技大学学报 2023年6期
朱华结,李志伟,贾云静,朱玉俊,Charles U.Pittman,Jr.
(1.河北科技大学化学与制药工程学院,河北石家庄 050018;2.河北大学生命科学学院,河北保定 071002;3.安徽医科大学药学院,安徽合肥 230012;4.密西西比州立大学化学系,Starkville,MS 39762)
化学是生命起源的源头,尤其是有机立体化学[1-4]。据报道,有很多种途径生成具有微小%ee值的L-氨基酸[5-13]。例如:由圆偏振光诱导的外消旋氨基酸的选择性分解D-氨基酸[14-16]、手性对称性破缺等[17-19]。实际上,有2种可能的途径来增加原始海洋(池)中的(R)-或(S)-对映异构体含量。一种是化学方法,另一种是物理途径。化学方法包括手性放大(扩增)[20-25],如在手性催化剂存在下将二乙基锌添加到醛中观察到的手性扩增[21],这也被称为非线性效应[22-24],其包含正非线性效应(不对称放大)和负非线性效应(非对称耗尽)。那些较小%ee1值的手性物质(如L-氨基酸),也可能由于自身催化(autocatalysis)[25]或者手性助剂的作用[20-24,26],最终的对映体含量得到提高。显然,非线性效应并不能保证那些具有较小%ee值的手性化合物的手性放大。另外,也有不需要手性催化剂来增加%ee值的研究。例如:通过在惰性气氛下加热固体丝氨酸的非外消旋样品至175~230 ℃的温度,可以富集丝氨酸的%ee值[27]。这也可能是一个方法,在原始海洋(池)中不存在任何其他手性催化剂的情况下,L-氨基酸的%ee值可以得到增加。(1)有关生命起源方面的研究论文有上千篇之多,由于篇幅限制,本文无法引用更多的文献,请读者理解。
物理方法包括利用D-和L-氨基酸的外消旋体的盐在水中较低溶解度而被分离出来。例如,相同摩尔的D-和L-氨基酸可以形成相应的盐晶体,这些盐晶体具有较低的溶解度。因此,如果L-氨基酸在溶液中有一点过量,这些晶体沉淀下来后,L-氨基酸的%ee值会得到提高[28]。事实上,作为最古老的问题之一,虽然有非常多关于生命起源的报道[29],但是人们的研究兴趣依然不减。(2)在使用HPLC进行手性对映体含量分析的过程中,其含量分析的误差主要来自相对误差,即分析过程中使用积分的时间区间。因此,在分析过程中,对每一对样品的分析,其积分区间的时间严格保持在几乎相等的时间段。在这种情况下,相对误差可以控制在最小的范围。本文中大部分%ee值的差异在1%以上,极少数在1%ee以下。因此,对于这部分极小的%ee差值,可能存在相对较大的误差。但是,纵观全部数据,可以看到其手性放大的大趋势变化与理论值的变化趋势一致。为了更直观地了解HPLC谱图的分析结果,将6个典型的原始HPLC及分析结果的截屏图总结在表2中。如果需要,读者可来信索要。本文在给《河北科技大学学报》投稿前给Molecules投稿的英文稿件和英文支持信息中有更多英文文献和详细的实验和计算结果,如绝对构型鉴定、不同立体构型异构体的能量、产物的理论产率、实验和计算的ECD光谱、OR数据等。
任何能提高L-氨基酸%ee值的理论和/或实验方法都值得重视。在史前时代、生命起源之前,低%ee值化合物的混合物可能存在并且持续存在了很长一段时间。因此,最初微小的%ee值的非外消旋化合物如何在某个阶段生成较高%ee值,并在进一步的演化过程中,生成相关的多肽等与生命起源有重要作用的物质,就构成了一个研究的重点内容。在有机化学反应中,例如使用手性催化剂催化二乙基锌与苯甲醛反应生成手性醇,其反应起始的%ee值与反应后产物的%ee值,与手性催化剂的%ee值等因素有直接的关系。这方面已经有不少报道[23]。然而,在没有手性催化剂作用下,那些小%ee值的非消旋化合物在通过某类反应后,其留存在溶液中的手性对映体%ee值的变化与反应中的哪些因素有关?基于早期的实验结果[30],本文将探讨提高具有较小%ee值的非消旋化合物的%ee值的理论方法,并在系列实验中证实了初始的%ee值(%ee1)和最终%ee值(%ee2)之间的关系。
1 实验部分和计算部分
1.1 实验部分
通过混合已知对映体含量的L-色氨酸甲酯(L-TME)和D-TME商业样品,制备出L-TME的%ee1值在1.0%左右。由于初始%ee1值非常小,因此采用了3种测定方法对%ee1进行测定。第1种方法直接测定其特征旋光(OR)值并将其与99%ee的L-TME样品的OR值进行比较。第2种方法是使用手性柱进行HPLC分析L-TME的含量。第3种方法是让L-TME与苯甲酰氯进行反应得到相应的苯甲酰胺,测量获得的(S)-苯甲酰氯的OR数值并用HPLC分析方法测定%ee1。例如:使用Chiralcel-IA柱用于分析其%ee值,用石油和异丙醇(体积比为60∶40)进行洗脱。这3种方法所得出的结果几乎相同。相关的HPLC分析和数据处理见注解②。
将5.0 g含量为0.7%~1.0%ee的L-TME的原料溶解在CH2Cl2中并将其冷却至接近0 ℃,加入丁二酮(其摩尔量是D-TME的90%)。将混合物搅拌10 h,加入0.01%三氟乙酸(TFA),然后加入少量分子筛(4Å),该Pictet-Spengler反应在0 ℃条件下反应14 h,然后回收未反应的原料。在相同反应条件下,将所回收的未反应的起始原料用于第2次的Pictet-Spengler反应(丁二酮与D-TME摩尔比为0.90∶1)。在第2次Pictet-Spengler反应后,回收未反应的原料,利用手性HPLC分析未反应物的%ee2值。通过2次Pictet-Spengler反应,%ee2/%ee1的值在2.7~3.2之间。
将具有不同%ee1值的L-TME样品与草酰氯反应,在零下80 ℃的条件下加入三乙胺(TEA)后,维持零下80 ℃反应24 h。反应结束后,将未反应的L-TME粗品通过硅胶柱进行纯化,然后通过手性HPLC分析回收纯TME样品的%ee值。
由于部分产物的%ee值较小,因此在使用HPLC分析过程中,需要对每个分析样品的积分区间进行严格控制。对于同一批次的样品分析,每一组的积分区间几乎都是相同的。使用石油醚和异丙醇(体积比为80∶20)进行淋洗。
1.2 计算部分
使用构象搜索软件对相关分子进行构象搜索,对相对能量为0~25 kJ/mol的构象进行再次计算(B3LYP/6-311++G(2d,P))。使用Gaussian09软件进行相关分子不同构型的计算,相对能量为零的结构为最优结构。通过计算该分子的电子圆二色谱并与实验值进行比较,鉴定相关分子的绝对构型(本文报道的结构即为已经鉴定出来的绝对构型)[31-32]。
2 结果与讨论
2.1 不含手性源手性富集/放大的理论研究
有研究表明,在加热条件下,没有反应的丝氨酸的对映体含量出现了明显增大(手性放大或富集)[27]。在使用乙二醛与L-TME进行反应时,发现L-TME倾向于与D-TME反应[30]。这表明在没有任何手性催化剂的情况下手性放大(富集)是有可行性的。因此,首先从数学上研究起始%ee值与最终%ee值之间的对应关系。
生命起源前的化合物中,最早形成的手性化合物(包括L-氨基酸)的%ee值会非常小(见图1)。在史前阶段,这些非常小%ee值的非外消旋化合物可能需要一种更有效和可靠的方法来提高%ee值,以便用于后期合成单一手性的多肽等物质。在第Ⅱ阶段,手性化合物具有确定的%ee值后,其%ee值的提高可能涉及各种方法,如手性放大或自催化等方法。在最后阶段,随着L-氨基酸含量的增加,可能形成相应的单一手性肽或其他用于生命起源的生物活性化合物,或者充当手性源用于其他手性化合物的合成。然而,第Ⅰ阶段(最早阶段)可能需要更可靠的方法来保证将%ee值提高到合理的水平,以便后期使用。本文报道一种可以提高手性化合物(如L-氨基酸)%ee值的普适性方法,该方法可以100%将其初始%ee值提高到一个合适的程度。

图1 具有小%ee值的非外消旋性手性化合物在生命起源前的史前时期3个可能阶段的手性富集/放大
例如,当一个(R)-M(1)及其对映体(S)-M(2)可以与含双官能团的反应物(bis-FG)反应(见方案1),其中丁二酮是双官能试剂。反应在0~5 ℃的二氯甲烷溶液中进行[33-34]。方案1中,TME与丁二酮反应后,其过量的(S)对映体(L-TME)留存在溶液中。反应生成化合物3(外消旋体3(1)和3(2))及化合物4(外消旋物(4(1)和4(2))[31,35-38]。分离得到的化合物3的产率为18%,化合物4的产率为35%,总产率为52%。通过计算其电子圆二色谱并与实验圆二色谱进行比较,鉴定得到化合物3和4的绝对构型[39-42]。该反应将进一步用于推导相关的方程,以确定反应后TME对映异构体的%ee2值与其初始%ee1之间的关系。
2.2 4种反应情况
在第1种情况下,一个分子的1和一个分子的2与bis-FG反应生成产物(R,S)-3(i) (i=1~n),这里i是基团化合物3的第i个产物,n是3的第n个产物。例如,第1个化合物(i=1),被定义为(R,S)-3(1);第2个化合物被定义为(R,S)-3(2);第2种情况,反应发生在1和2与bis-FG反应生成(S,R)-3(i)。这样,(R,S)-3(i)是其对映体。第3种情况是2个分子1与一个bis-FG反应生成化合物(R,R)-4(j) (j=1~m),其中j表示4的第j个产物,m是4最后一个化合物。在第4种情况中,2的2个分子与bis-FG反应生成(S,S)-4(j) (j=1~m)。因此,这4种情况下,产物3的总数为(R,S)-3(i)和(S,R)-3(i)的总和。同样,4的所有化合物数量也是如此。见方案2。
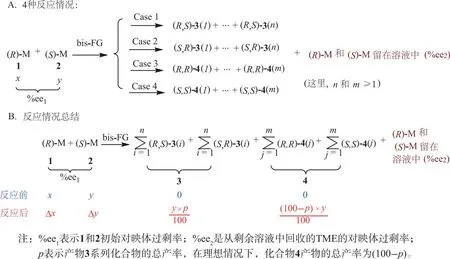
方案2 4种反应情况的简化表达式

化合物4生成的总量为 (100-p)×y/100,这样,Δx和Δy似乎可以在3和4形成后,按照下式计算:
(1)
(2)
这样,其最终%ee2为
(3)
这显然表明%ee2与产率 (p) 和3无关。但是,这个推导出来的式(3)是错误的。这是因为反应中生成的(R,R)-4和(S,S)-4的量不同。由于生成(R,R)-4和(S,S)-4具有相同的过渡态能垒,因此生成的(R,R)-4和(S,S)-4的数量一定取决于3生成后溶液中剩余的1和2的数量之比。因此,当初始x>y时,1和2的消耗量与生成3的数量相同。这样,Δx一定大于Δy。生成(R,R)-4的量不等于(S,S)-4。1和2在4的形成过程中,所消耗的量不同。因此在这种情况下,用4的总量[(100-p)×y/100]代替式(2)和式(3)中的1和2的量来计算Δx和Δy不正确。下面推导在形成3后溶液中剩余的1和2的量。
形成(y×p/100)摩尔3后,生成4的数量为(100-p)×y/100。由于(R,R)-和(S,S)-4系列化合物必须有相同的过渡态垒,因此,溶液中(R,R)-4与(S,S)-4的比值取决于溶液中剩余的1和2的比值。也就是说,溶液中(R,R)-4与(S,S)-4比例和溶液中1和2的数量成正比。如果1多,则(R,R)-4生成数量多。因此,在(y×p/100)摩尔3形成后,1消耗掉QR,R用于4的合成,可表示为式(4)。
(4)
同样,2用于合成系列(S,S)-4的数量为QS,S。
(5)
这样,生成3和4后,1和2剩余量分别为Δx和Δy:
(6)
(7)
因此,1在溶液中剩余的对映体量(%ee2)可以用式(8)计算:
(8)
将x和y代入式(8)后,可用下式计算:
(8A)
它与%ee1的关系也可以表示为
(8B)
因此,对于任意初始值%ee1,%ee2主要是化合物3产率(p)的函数。
2.3 x,y和p之间的关系
这里讨论由式(8)得出的3个主要特征。图2 a)为1的%ee2与%ee1的关系,图2 b)表明%ee值的增量Δ%ee=%ee2-%ee1与%ee1的关系,图2 c)描述了 %ee2与%ee1比值与%ee1的关系。分别计算了9种不同产率(p)时%ee的变化情况,从10%开始,每步增量为10%,直到90%。
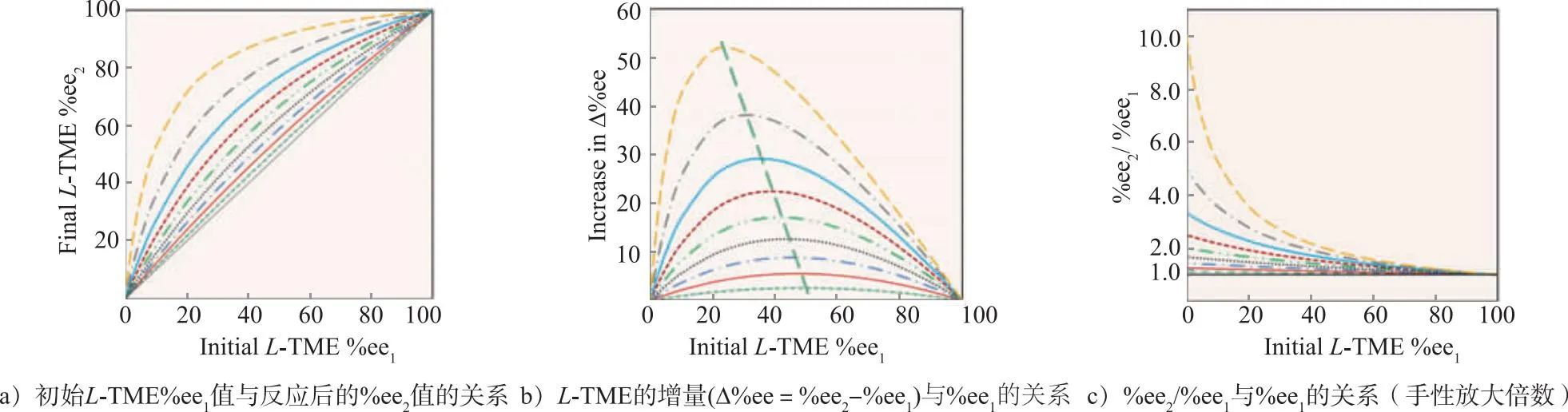
图2 %ee1,%ee2,Δ%ee与%ee2/%ee1之间的关系
图2 a)中所有9条%ee1vs %ee2曲线位于45°对角线(黑线)之上,3的产率(p)越高,溶液中的对映体增量Δ%ee就越大;图2 b)表明,3的产率越大,达到Δ%ee的最大增量所需的初始%ee1就越小;图2 c)显示富集/放大倍数不是线性的,%ee1值越小,%ee2/%ee1比值富集/放大倍数就越大,当p接近90%时,放大倍数为10。为了更清楚地说明p在大于95%时%ee1,以及Δ%ee和%ee2/%ee1之间的关系,本文计算并绘出了相关联系,见图3。从最低到最高的4条曲线分别表示产率(p)从95%,97%,99%到99.99%的变化情况。

图3 基于式(8A)的3种理论关系(式(8A)中的x>y)
当产率(p)从95%变化到99.99%时,且当%ee1为极小值0.005%时,其理论上的放大倍数分别接近20,33,99和6 667。当产率接近100%时,则放大倍数%ee2/%ee1达到20 000(见图3 c),限定y轴为50)。初始%ee1越低,放大倍数越大。这表明了一种极其重要的可能性,即1具有极小初始值%ee1时,都可能与bis-FG试剂反应,并快速提高其%ee2的值。
2.4 手性富集(放大)实验
2.4.1 使用TME和2,3-丁二酮反应进行的手性富集/放大
对映体过量的L-氨基酸或其酯对生命起源具有重要意义。为此,选用D-TME (1)和L-TME(2)作为手性原料,丁二酮作为双功能试剂,参见方案3。3的合成与4的合成是竞争性反应,因为中间体羟基胺在亚胺形成和环化发生之前与试剂处于平衡。化合物3生成的越多,溶液中L-TME的%ee2值就越高。因此,反应中的平衡时间分别选择2,10,36 h。反应结束后,分别测量溶液中L-TME的%ee2值,如图4所示。可以看到,所有实验得到的L-%ee2值都处于传统手性放大的区域。根据不同的理论计算方法,得到化合物3和4之间的能量差值,由此计算出3的相对理论产率(p值分别为21.7%,31.2%,33.3%)。将这些产率代入式(8)计算得到相应的%ee2。实验数据用方形、三角形、叉号绘制。
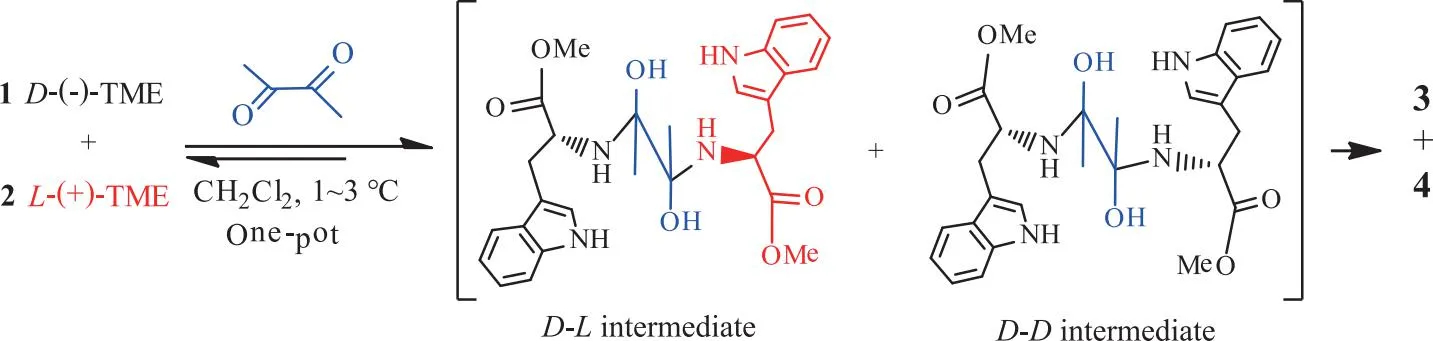
方案3 对映体1和2与丁二酮反应生成产物3和4
初始%ee1值与反应后的%ee2值及其增量等实验数据见表1。
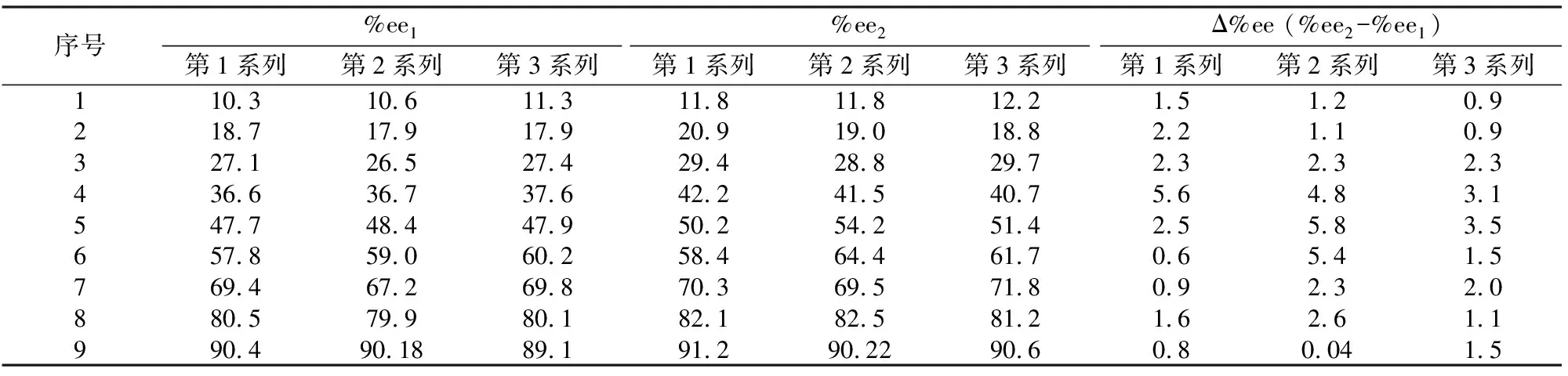
表1 初始%ee1值与反应后的%ee2值以及其增量等实验数据*
在这个反应后,L-TME的%ee值得到了提高,观察到的手性富集(放大)是该反应的必然结果。采用量子化学计算方法,分别计算3和4的最稳定构象,并分别计算出二者的相对能量。例如,计算采用B3LYP理论,使用6-311+G(d)和6-311++G(2d,p)基组,在气相或者在二氯甲烷中使用PCM模型进行计算[36-42]。计算表明,4比3更稳定,二者之间的能量差分别为2.174,1.906,2.829 kJ/mol。由此计算得到3的产率为21.7%或31.2%或33.3%。实验测定的L-TME对映体过剩率大部分位于计算得到产率为21.7%的曲线附近(见图4 b))。这些实验结果证实,L-和D-TME溶液中较低的最初始%ee1值在反应后得到了提高(%ee2值增大)。
2.4.2L-TME的初始0.7%ee提高到大约3%ee
配置了初始%ee1在0.7~1.0%ee之间的样品,可使用OR或者通过HPLC方法测定其%ee1值。第1次反应后,其放大倍数(%ee2/%ee1)为1.3~1.4。在回收未反应的样品并再次进行反应后,采用不同方法进行测定,得到L-TME的%ee2值在2.7~3.3之间,其放大倍数平均约为3.5。
用11.50 g的0.8%eeL-TME重复该过程,采用HPLC分析发现其反应后的溶液中产物的%ee2值为3.3,使用OR的数据为3.0%ee。该%ee2值比原始%ee1值大了约4.2倍,与使用5.00 gL-TME的第1次试验中获得的结果(平均值3.5)接近。
2.4.3 使用乙二醛与L-TME反应
在前述反应中,通过2次Pictet-Spengler反应可提高%ee值,但是%ee值的增量并不是太大。原因可能是丁二酮的2个甲基的空间位阻导致3的生成产率偏低。为此,进一步用空间位阻小的乙二醛进行反应(3)乙二醛在与TME反应中,由于商品乙二醛中含有少量甲醛( 0.5%),而甲醛也能与TME反应,关键问题是该产物的极性与TME极为接近。因此在回收的反应TME中,含有少量该产物。而这影响了回收TME的纯度,不方便用于进一步反应。尤其在配置1%ee或以下的非外消旋样品用于2次或以上的Pictet-Spengler反应时,最后发现回收的产物全是甲醛与TME的反应产物。因此配置小%ee值的样品用于多次Pictet-Spengler反应,不适合用乙二醛作双功能基试剂。(见方案4),生成了化合物5(2个外消旋体5(1)和5(2))和6(1个外消旋体6(1))。理论计算得到5(1)的相对能量低于6(1)的相对能量(5.47 kJ/mol)(计算在B3LYP/6-311++G(2d,p)基组上进行(PCM模型,溶剂是CH2Cl2))(4)通常,为了测定3和4在反应中的比例,通常要计算化合物3和4在形成前的中间体不同构象的能量差。然而,由于构象实在太多,且也需要考虑不同构型的构象分布,因此,一个简化的处理方法就是计算化合物3和4不同构型的相对能量。该化合物有64种不同的可能构型,因此,所有的构型一半,即所有的32个对映体的构型用于分析,采用 FFMM94S 分子力场进行构象分析。然后,采用2种不同的量子计算方法用于计算。从所有的计算中,获得最低的和次低的2个稳定构型结构用于分析。化合物的绝对构型鉴定的详细信息,可来信免费提供。。同时利用这个能量差数据和式(8)进行理论计算得到%ee2值并与实验值进行对比,如图5所示。

方案4 L-TME(%ee1)与乙二醛反应得到5(1),5(2)和6(1)
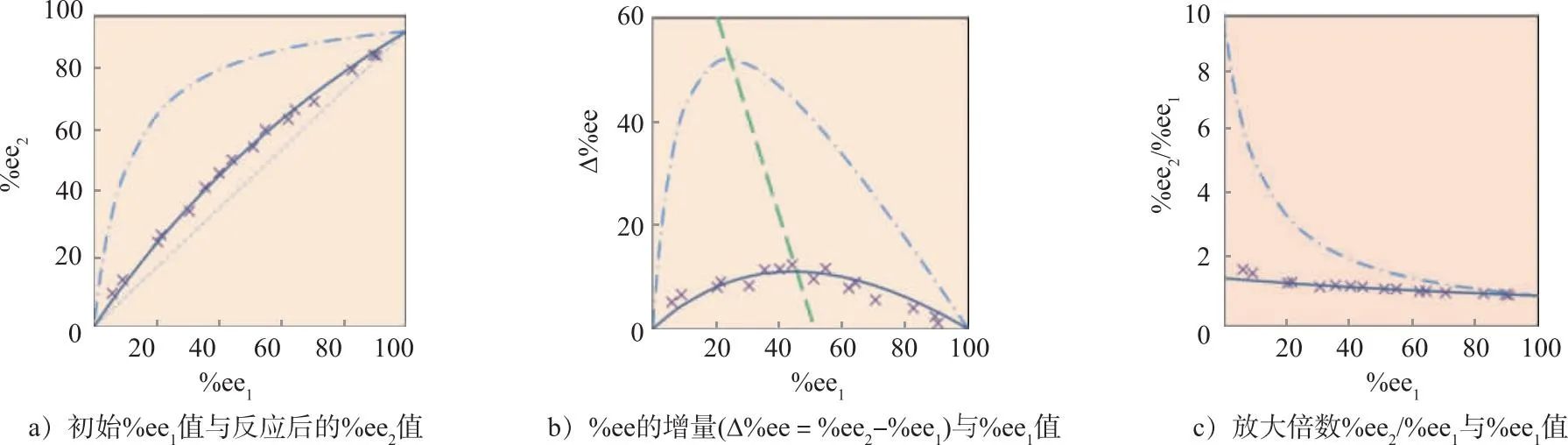
图5 方案4中反应时%ee1,%ee2,Δ%ee与%ee2/%ee1之间的关系(图中虚线由式(8)计算得到,其中的产率p由计算二者的能量差(5.47 kJ/mol)得到)
利用式(8)计算%ee2值随%ee1值的变化关系并与实验值进行比较,其中以5(1)和6(1)之间5.463 kJ/mol的能量差计算得到(R,S)-5的产率p为90%,符号“×”表示实验值。
乙二醛与D-TME的摩尔比为0.90∶1,L-TME的最初%ee1值从6.1%增加到89.6%。典型的HPLC分析图列于表2(为清晰起见,仅将对映体的2个信号附近区间截图),发现最大的放大倍数是1.92(参见表3)。当%ee1值变为89.6%时,放大倍数仅为1.03。%ee1越小,放大倍数越大。%ee增量最大值为12.3%。大多数%ee1值从将近22%到64%时,其增量在10%左右。
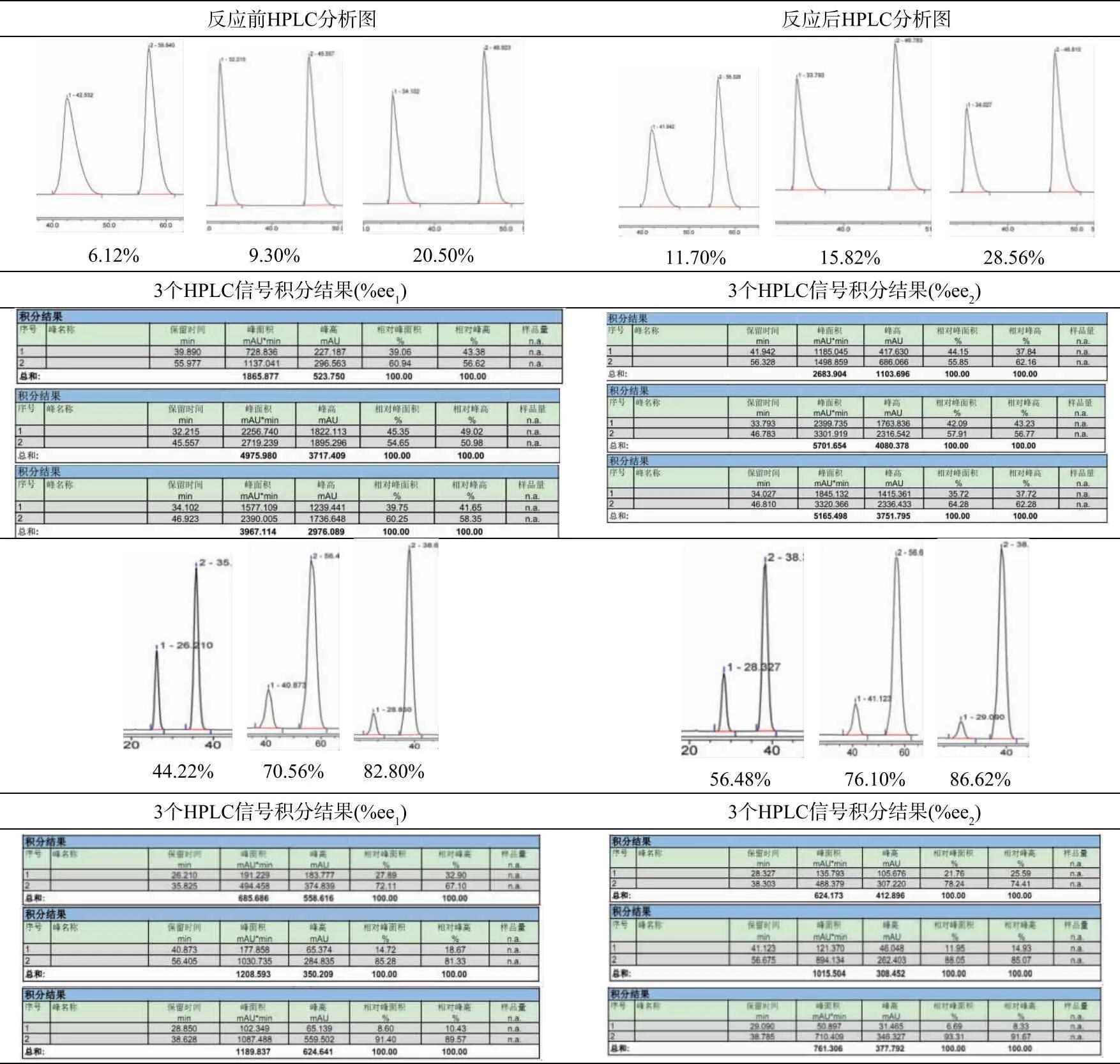
表2 不同浓度下测试的HPLC谱图和反应后回收物质的谱图截图以及分析结果(总结在表3)*
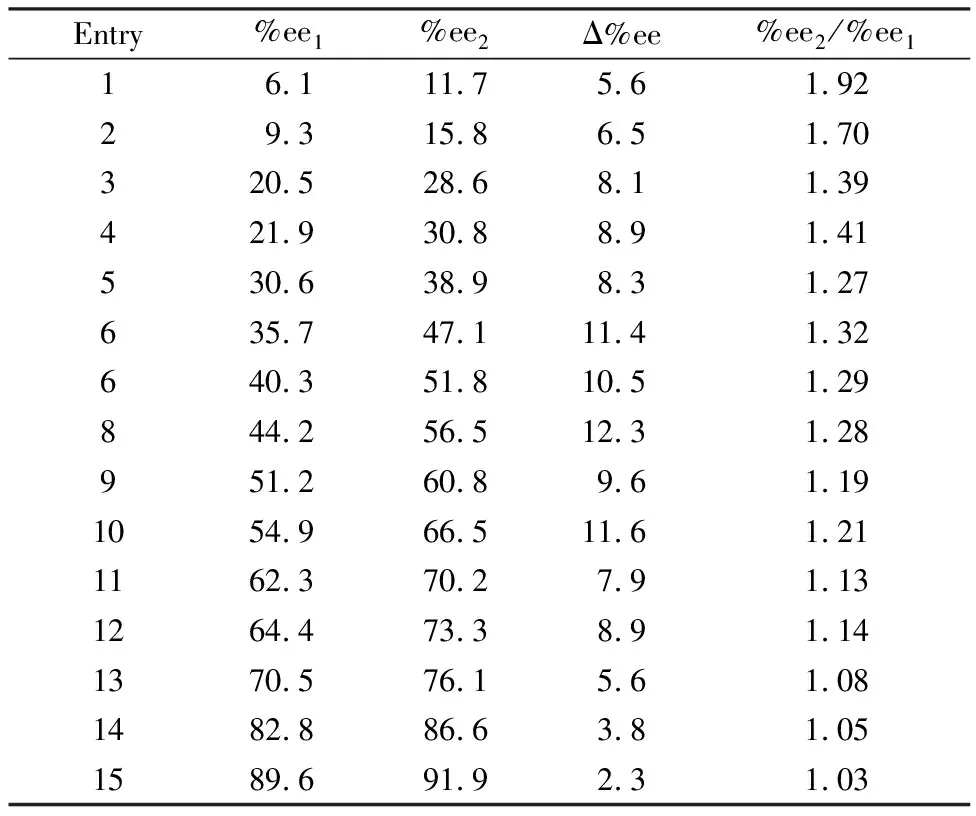
表3 L-TME在方案4反应中%ee值的富集/放大(部分HPLC分析截图见表2)*
最后,将30.6%ee的L-TME与不同比例的乙二醛/D-TME(摩尔比为0.7~1.5)反应,得到%ee2值与不同摩尔比的实验结果(见图6)。随着乙二醛与D-TME比例的增加,%ee2变化值从5.0%上升到12.1%,见表4。
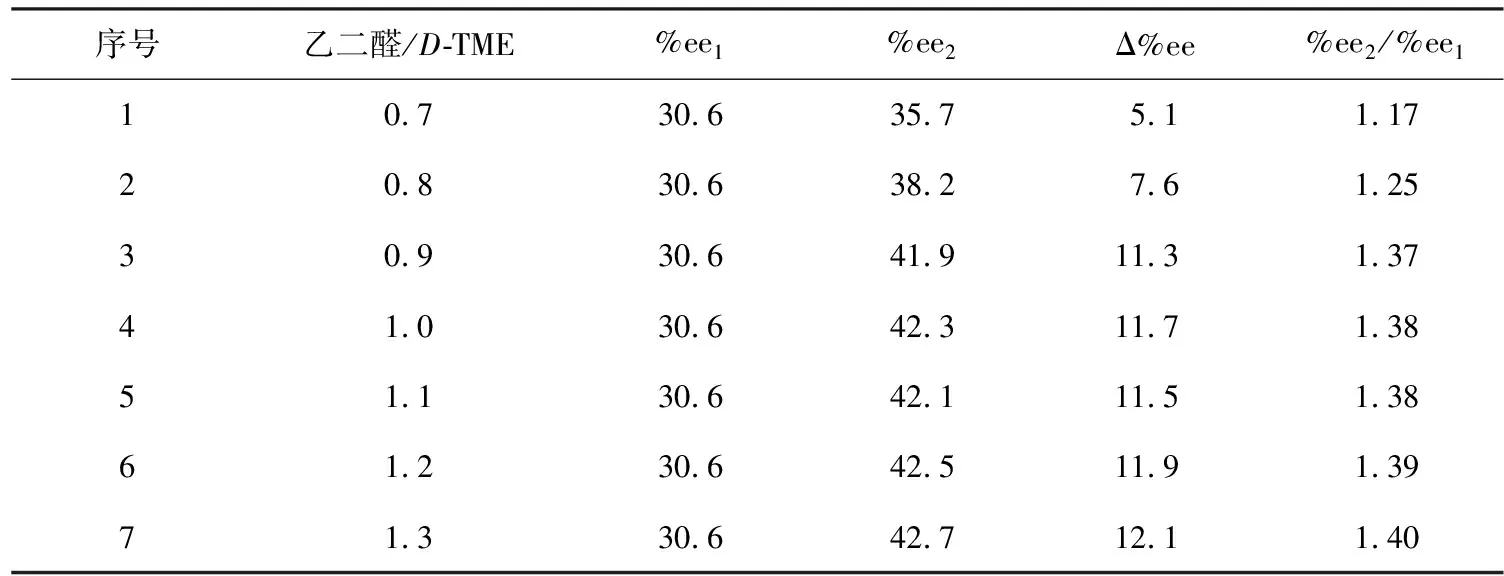
表4 起初原料%ee1值不变且乙二醛与D-TME物质的量比从0.7提高到1.3时,%ee2值变化趋势
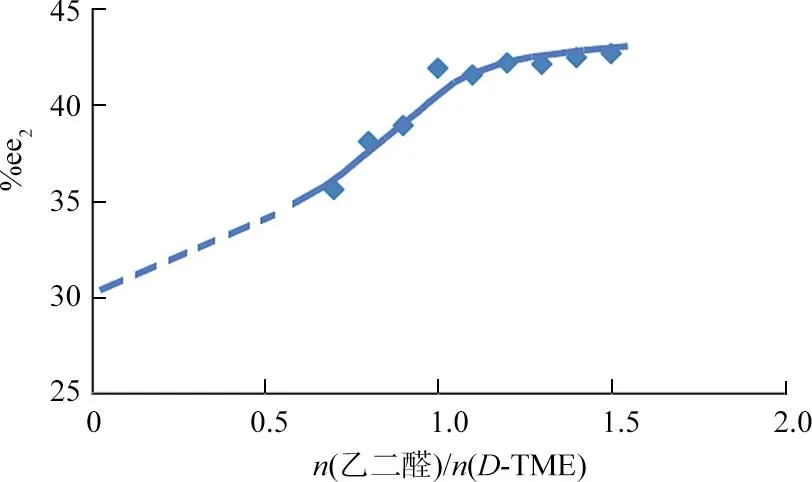
图6 反应后原料的%ee2值随乙二醛与D-TME摩尔比变化情况(%ee1=30.6%)
2.4.4L-苯丙氨酸手性富集
前面研究了丁二酮与TME反应中对映体过剩率的富集/放大问题。然而,在非平衡条件和低温下,这个结论是否依旧成立呢?为此,本文选择草酰氯作为双官能团试剂(见式(9))。在零下80 ℃温度下,与三乙胺(TEA)共存24 h。当L-苯丙氨酸甲酯的%ee1值从22.0%变为26.8%,46.6%和56.0%时,L-苯丙氨酸甲酯%ee值的增加量(Δ%ee)分别为1.8%,2.3%,3.9%和0.8%。由于草酰氯在TEA催化下具有非常高的反应活性,因此,当63.9%的L-苯丙氨酸(%ee1)用于反应时,%ee值的增加量(Δ%ee)仅仅为0.3%。
(9)
2.4.5 通过环状二肽二聚富集L-氨基酸
氨基酸可能也参与了生命起源前的缩合反应生成环肽。其中L-氨基酸与另一种L-或D-氨基酸反应,得到相应的L,L-和D,L-环二肽(见式(10))。剩余未聚合的L-氨基酸,如果最初以微小的%ee1存在,反应后可能会被放大到更高的%ee。选择6种典型的氨基酸进行计算,研究%ee的放大,分别是最小的手性氨基酸丙氨酸、含有双立体中心的异亮氨酸、带有异丙基的缬氨酸、含有硫醇的半胱氨酸、带有杂环部分的组氨酸和带有2个羧酸基团的天冬氨酸。采用B3LYP和MPW1PW91理论方法在6-311++G(2d,p)基组上计算在水中(PCM模型)的能量,得到L,L-和D,L-环二聚体的能量差,用于计算环二聚体产率(p)下,%ee1=0.50%时的L-氨基酸值的%ee2值、增加量(Δ%ee)和%ee2/%ee1的值(见表5)[43]。

表5 6个氨基酸形成的L,L-和D,L-环二肽之间的能量差及用该数据计算得到的%ee2和Δ%ee值
(10)
由表5可知,半胱氨酸(17)和组氨酸(19),在水中形成的D,L-环二肽比它们在水中对应的L,L-产物具有更低的能量。例如,D,L-产物分别降低0.075和2.616 kJ/mol。半胱氨酸形成的环二肽结构中未发现分子内氢键。但是,在仔细检查这些低能量D,L-环二肽几何结构后,发现组氨酸的最低能量结构中有分子内氢键,而水很可能通过溶剂化作用破坏分子内氢键。因此,进一步选择有没有分子内氢键的构象来进行能量分析。
当能量计算中不包含具有氢键的几何结构时,组氨酸酯的D,L-环二聚体的能量比L,L-产物低4.288 kJ/mol。类似地,天冬氨酸酯在这些条件下的D,L-和L,L-产物之间的能量差为2.286 kJ/mol。因此,如果所有原始氨基酸都转化为L,L-和D,L-环肽(见式(10)),则形成的最多产物的产率(在D-对映体完全转化时)将取决于2种产物之间的相对能量。环二聚体产物的相对能量越高,该产物的产率就越小。大多数D,L-环二聚体的能量低于它们的L,L-产物的能量。因此,L-和D-氨基酸倾向于形成D,L-环二肽为主要产物。形成的D,L产物越多,溶液中未反应的L-氨基酸就越多。根据B3LYP计算的能量,得到式(10)中D,L-环二肽的产率(p)(对应于(R,S)-产物),%ee1,%ee2,Δ%ee和放大倍数%ee2/%ee1之间的关系(利用式8)),见表4。理论上,剩余L-氨基酸的%ee值将增加。
2.5 相关化合物数据
化合物3(1):(6R,7aR,12bS,14S,15aS,15bR)-15a,15b-dimethyl-5,6,7a,8,13,14,15,15a,15b,16-decahydropyrrolo[2′,3′:1,2]indolizino[3,2-b:8,7-b′]diindole-6,14-dicarboxylate。[α]D+32 (c3.25,CH2Cl2)。HR-MS-ESIm/z,计算得到 C28H30N4O4[M+H]+487.234 5,实验值为487.235 6。1H NMR (600 MHz,CDCl3)δ9.85(s,1H),7.39(d,J=7.8 Hz,1H),7.31(d,J=7.86 Hz,1H),7.05(t,J=7.2 Hz,1H),6.99(m,2H),6.88(d,J=7.2 Hz,1H),6.63(t,J=7.2 Hz,1H),6.49(d,J=8.4 Hz,1H),5.10(s,1H),4.45(dd,J=12,7.2 Hz,1H),4.12(dd,J=12.0,5.1 Hz,1H),3.76(s,3H),3.39(s,3H),3.27(dd,J=15.6,12.0 Hz,1H),2.88(dd,J=15.6,4.8 Hz,1H),2.15(m,1H),1.38(s,3H),1.30(m,1H),1.11(s,3H)。13C NMR(150 MHz,CDCl3)δ173.4,172.8,149.2,138.2,135.4,129.9,127.5,125.7,122.8,120.4,118.0,116.8,116.6,110.7,108.2,104.5 80.0,74.2,66.9,63.7,60.8,51.3,51.0,42.0,23.3,21.1,19.2。
化合物4(2):(6S,7aS,12bR,14S,15aR,15bR)-15a,15b-dimethyl-5,6,7a,8,13,14,15,15a,15b,16-decahydropyrrolo[2′,3′:1,2]indolizino[3,2-b:8,7-b′]diindole-6,14-dicarboxylate。[α]D-104.76(c,5.25,CHCl3)。HR-MS-ESIm/z,计算得到 C28H30N4O4[M+H]+487.234 5,实验值为487.235 4。1H NMR(600 MHz,CDCl3) δ 7.88(s,1H),7.44(d,J=7.8 Hz,1 H,),7.24(d,J=7.8 Hz,1 H),7.06(m,2 H,),7.02(m,2 H),6.65(t,J=7.2 Hz,1 H,),6.54(d,J=7.8 Hz,1 H,),4.78(s,1 H),4.29(t,J=5.4 Hz,1 H),3.89(dd,J=10.2,4.8 Hz,1 H),3.62(s,3 H),3.58(s,3 H),3.27(dd,J=15,6.0 Hz,1 H,),3.12(dd,J= 15,5.4 Hz,1 H),2.51~2.41(dd,J=14.4,4.8 Hz,2 H),1.43(s,3 H),1.04(s,3H)。13C NMR(150 MHz,CDCl3) δ 175.5,175.0,150.7,136.3,136.0,129.7,128.7,126.6,124.1,122.0,119.6,118.4,118.0,111.2,109.7,107.8,89.7,77.7,66.6,65.3,59.6,57.3,52.3,52.2,43.3,26.2,26.2,23.4。
在%ee值分析中,回收的色氨酸甲酯需要与苯甲酰氯反应生成相应的苯甲酰胺,用于分析其%ee2。该苯甲酰胺的NMR数据:1H NMR(600 MHz,CDCl3)δ8.26(s,1 H),7.68(d,J= 7.9 Hz,2 H),7.55(d,J= 7.9 Hz,1 H),7.47(t,J= 7.4 Hz,1 H),7.40~7.32(m,3 H),7.18(t,J= 7.6 Hz,1 H),7.08(t,J= 7.5 Hz,1 H),6.99(d,J= 2.1 Hz,1 H),6.69(d,J= 7.4 Hz,1 H),5.15(dt,J= 7.5,5.3 Hz,1 H),3.71(s,3 H),3.45(t,J= 5.8 Hz,2 H)。13C NMR(151 MHz,CDCl3)δ172.42(s),166.96(s),136.17(s),133.89(s),131.71(s),128.55(s),127.69(s),127.09(s),122.84(s),122.30(s),119.74(s),118.68(s),111.32(s),110.06(s),53.50(s),52.43(s),27.69(s)。13C NMR(151 MHz,CDCl3) δ 172.42(s),166.96(s),136.17(s),133.89(s),131.71(s),128.55(s),127.69(s),127.09(s),122.84(s),122.30(s),119.74(s),118.68(s),111.32(s),110.06(s),53.50(s),52.43(s),27.69(s)。
3 结 语
通过数学理论推导结合有机反应原理,发现在没有手性催化剂或额外手性试剂存在的情况下,可以实现将较低%ee值的任何对映体的过剩率通过合适的化学反应来实现放大。实验研究证实了这些结论。采用这种实验方法,将低%ee值的手性对映体含量提高(超过10%ee或更高)后,这些%ee值高的对映体可以作为种子在生命起源前的原始海洋(池)内促进或催化其他手性化合物的合成,例如,L-氨基酸催化D-糖的合成。
生命的诞生说明在自然界中手性分子的相互作用有其自身的规律。概率论的结论是生命来自偶然。但是本文中的数学公式说明,哪怕L-氨基酸稍微过量一点,数学和化学的原则就保证了这些稍微过量一点的L-氨基酸能够在温和的条件下(这也是生命起源所需要的),其%ee值可以得到有效提高。这与非线性效应(non-linear effect)的结论完全不同(手性可能放大也可能被耗尽)。最新报道的实验和理论计算也表明:L-氨基酸能催化D型四碳糖合成,而L-氨基酸和D-糖恰恰是构成生命最基本的两类物质。因此,生命起源的过程中,概率论也许是其中的一个可能性。但是本文的研究表明,手性的放大是化学上的必然事件。虽然现在有关生命起源(手性放大/富集)中化学部分的研究已经不再是某些杂志的热点,但是对其深入研究,挖掘其背后的必然性,在生命起源研究中发现更多的必然因素和规律,也许是化学家、物理学家和生物学家,包括天文学家的使命之一。
致谢:感谢河北大学超级计算中心给予计算支持;感谢中科院北京微生物所李二伟博士、任俊伟博士提供部分NMR分析和旋光测量。
参考文献/References:
[1] WALDEP.Prebiotic Chemistry:From Simple Amphiphiles to Model Protocells[M].Berlin,Heidelberg:Springer,2005.
[2] DAS K,GABRIELLI L,PRINS L J.Chemically fueled self-assembly in biology and chemistry[J].Angewandte Chemie (International ed.in English),2021,60(37):20120-20143.
[3] DAVANKOV V A.Chance and necessity in the evolution of matter to life:a comprehensive hypothesis[J].Symmetry,2021,13(10).DOI:10.3390/sym13101918.
[4] JIA T Z,NISHIKAWA S,FUJISHIMA K.Sequencing the origins of life[J].BBA Advances,2022,2.DOI:10.1016/j.bbadva.2022.100049.
[5] PIZZARELLO S,GROY T L.Molecular asymmetry in extraterrestrial organic chemistry:An analytical perspective[J].Geochimica et Cosmochimica Acta,2011,75(2):645-656.
[6] KVENVOLDEN K,LAWLESS J,PERING K,et al.Evidence for extraterrestrial amino-acids and hydrocarbons in the Murchison meteorite[J].Nature,1970,228(5275):923-926.
[7] CRONIN J R,PIZZARELLO S.Enantiomeric excesses in meteoritic amino acids[J].Science,1997,275(5302):951-955.
[8] CRONIN J R,PIZZARELLO S,YUEN G U.Amino acids of the Murchison meteorite:II.Five carbon acyclic primaryβ-,γ-,and delta-amino alkanoic acids[J].Geochimica et Cosmochimica Acta,1985,49:2259-2265.
[9] PIZZARELLO S,KRISHNAMURTHY R V,EPSTEIN S,et al.Isotopic analyses of amino acids from the Murchison meteorite[J].Geochimica et Cosmochimica Acta,1991,55:905-910.
[10] PIZZARELLO S,WEBER A L.Prebiotic amino acids as asymmetric catalysts[J].Science,2004,303(5661).DOI:10.1126/science.1093057.
[11] LEVINE M,KENESKY C S,MAZORI D,et al.Enantioselective synthesis and enantiomeric amplification of amino acids under prebiotic conditions[J].Organic Letters,2008,10(12):2433-2436.
[12] BRESLOW R.A likely possible origin of homochirality in amino acids and sugars on prebiotic earth[J].Tetrahedron Letters,2011,52(17):2028-2032.
[13] GREENWALD J,KWIATKOWSKI W,RIEK R.Peptide amyloids in the origin of life[J].Journal of Molecular Biology,2018,430(20):3735-3750.
[14] FLORES J J,BONNER W A,MASSEY G A.Asymmetric photolysis of (RS)-leucine with circularly polarized ultraviolet light[J].Journal of the American Chemical Society,1977,99(11):3622-3625.
[15] BAILEY J,CHRYSOSTOMOU A,HOUGH J H,et al.Circular polarization in star-formation regions:implications for biomolecular homochirality[J].Science,1998,281(5377):672-674.
[16] RUBENSTEIN E,BONNER W A,NOYES H P,et al.Supernovae and life[J].Nature,1983,306(5939):118-118.
[17] MAUKSCH M,TSOGOEVA S B,WEI Shengwei,et al.Demonstration of spontaneous chiral symmetry breaking in asymmetric Mannich and Aldol reactions[J].Chirality,2007,19(10):816-825.
[18] VIEDMA C.Chiral symmetry breaking and complete chiral purity by thermodynamic-kinetic feedback near equilibrium:implications for the origin of biochirality[J].Astrobiology,2007,7(2):312-319.
[19] WEISSBUCH I,LEISEROWITZ L,Lahav M.Stochastic “Mirror Symmetry Breaking” via Self-Assembly,Reactivity and Amplification of Chirality:Relevance to Abiotic Conditions[M].Berlin Heidelberg:Springer,2005:123-165.
[20] KITAMURA M,SUGA S,KAWAI K,et al.Catalytic asymmetric induction:Highly enantioselective addition of dialkylzincs to aldehydes[J].Journal of the American Chemical Society,1986,108(19):6071-6072.
[21] KITAMURA M,SUGA S,OKA H,et al.Quantitative analysis of the chiral amplification in the amino alcohol-promoted asymmetric alkylation of aldehydes with dialkylzincs[J].Journal of the American Chemical Society,1998,120(38):9800-9809.
[22] SATYANARAYANA T,ABRAHAM S,KAGAN H B.Nonlinear effects in asymmetric catalysis[J].Angewandte Chemie International Edition,2009,48(3):456-494.
[23] GIRARD C,KAGAN H B.Nonlinear effects in asymmetric synthesis and stereoselective reactions:ten years of investigation[J].Angewandte Chemie,1998,37(21):2922-2959.
[24] PUCHOT C,SAMUEL O,DUNACH E,et al.Nonlinear effects in asymmetric synthesis:Examples in asymmetric oxidations and aldolization reactions[J].Journal of the American Chemical Society,1986,108(9):2353-2357.
[25] SOAI K,SHIBATA T,MORIOKA H,et al.Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule[J].Nature,1995,378(6559):767-768.
[26] BLACKMOND D G,KLUSSMANN M.Spoilt for choice:assessing phase behavior models for the evolution of homochirality[J].Chemical Communications,2007(39):3990-3996.
[27] PERRY R H,WU Chunping,NEFLIU M,et al.Serine sublimes with spontaneous chiral amplification[J].Chemical Communications,2007(10):1071-1073.
[28] THIEMANN W,WAGENER K.Is there energy difference between enantiomorphic structures?[J].Angewandte Chem,1970,9(9):740-741.
[29] RUIZ-MIRAZO K,BRIONES C,de la ESCOSURA A.Prebiotic systems chemistry:New perspectives for the origins of life[J].Chemical Reviews,2014,114(1):285-366.
[30] BAI Bing,LI Dashan,HUANG Shengzhou,et al.Chirality pairing recognition,a unique reaction forming spiral alkaloids from amino acids stereoselectively in one-pot[J].Natural Products and Bioprospecting,2012,2:53-58.
[31] 朱华结.手性药物分子绝对构型鉴定的理论研究进展[J].河北科技大学学报,2022,43(4),401-414.
ZHU Huajie.Progress of the theoretical research on absolute configuration assignment for chiral medicines[J].Journal of Hebei University of Science and Technology,2022,43(4):401-414.
[32] ZHU Huajia,WANG Yufeng,NAFIE L A.Computational methods and points for attention in absolute configuration determination[J].Frontiers in Natural Products,2023,DOI:10.3389/fntpr.2022.1086897.
[33] MUNDY B P,ELLERD M G,FAVALORO F G.NameReactions and Reagents in Organic Synthesis[M].Hoboken:Wiley,2005.
[34] BAILEY P D,COLLIER L D,HOLLINSHEAD S P,et al.A new asymmetric route to bridged indole alkaloids:Formal syntheses of (-)-suaveoline,(-)-raumacline and(-)-Nb-methylraumacline[J].Journal of the Chemical Society,Chemical Communications,1994(13):1559-1560.
[35] FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 03 User’s Reference[M].Carnegie:Gaussian Inc,2003.
[36] BEROVA N,POLAVARAPU P L,NAKANISH K,et al.Comprehensive Chiroptical Spectroscopy[M].Hoboken:John Wiley &Sons Inc,2011.
[37] ZHU Huajie.Organic Stereochemistry[M].Weinheim:Wiley-VCH Verlag GmbH &Co.KGaA,2015.
[38] HALGREN T A.MMFF V I.MMFF94s option for energy minimization studies[J].Journal of Computational Chemistry,1999,20(7):720-729.
[39] JORGENSEN P,JORGEN H,JENSEN A,et al.Linear response calculations for large scale multiconfiguration self-consistent field wave functions[J].The Journal of Chemical Physics,1988,89(6):3654-3661.
[40] AUTSCHBACH J,ZIEGLER T,van GISBERGEN S J A,et al.Chiroptical properties from time-dependent density functional theory (Ⅰ):Circular dichroism spectra of organic molecules[J].The Journal of Chemical Physics,2002,116(16):6930-6940.
[41] BAK K L,HANSEN A E,RUUD K,et al.Ab initio calculation of electronic circular dichroism fortrans-cyclooctene using London atomic orbitals[J].Theoretica ChimicaActa,1995,90:441-458.
[42] KONDRU P K,WIPF P,BERATAN D N.Theory-Assisted determination of absolute stereochemistry for complex natural products via computation of molar rotation angles[J].Journal of the American Chemical Society,1998,120(9):2204-2205.
[43] ZHAO Dan,ZHAO Qiqi,ZHU Huajie,et al.Theoretical investigation of the relationship between four-carbon d-sugars and fiveL-amino acids[J].Tetrahedron,2016,72(35):5558-5562.
猜你喜欢
中国环境科学(2023年10期)2023-10-26
分子催化(2022年1期)2022-11-02
林产工业(2022年10期)2022-10-30
云南化工(2021年8期)2021-12-21
闽南师范大学学报(自然科学版)(2020年2期)2020-07-06
中成药(2017年9期)2017-12-19
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
分析测试学报(2015年8期)2016-01-13
郑州大学学报(理学版)(2014年3期)2014-03-01
