
以基因定位为主线的遗传学教学构架及案例
2023-12-21 03:09周希希贺乔乔张晓娟
生物学杂志 2023年6期
张 羽,王 瑀,周希希,吴 栋,贺乔乔,张晓娟
(陕西理工大学 生物科学与工程学院,汉中 723000)
21世纪生命科学表现出前所未有的发展势头,成为自然科学的带头学科之一,其中,人类对遗传物质本质和生命奥秘的探索,构成了生命科学进步的支柱,因此,遗传学成为生命科学的引领学科[1]。近年来,生物技术迅猛发展,特别是测序技术广泛应用,使遗传学课程的教学资源不断丰富,国内外主要出版的遗传学教材已达十几种,其课程内容体系构建各不相同[2-5],其中,许多版本教材的内容多达20余章,涉及知识面非常广泛;目前,遗传学教学面临如下问题和挑战:涉及分子层面的内容越来越多、抽象性越来越强;生物学大数据喷涌式释放;交叉学科知识不断增加;学时不断减少等,导致很多学生认为遗传学是生命科学领域最难的一门课程,在学习中产生畏难情绪。遗传学教改论文多注重教学手段和方法探索[6-15]。针对遗传学的主要研究内容,设计一条主线突出的教案,可为学生的自主和探究式学习提供很好的抓手,同时教案在教学过程中起到总体导向、规划和指导作用。在遗传学课程中,对基因本质的认识和基因定位一直是遗传学中研究的主要内容,本文用知识导图和思维导图的形式设计几个“基因定位”的教学案例,旨在为提高遗传学教学质量和效果,打造遗传学一流课程,培养生命科学领域高素质人才提供借鉴。
1 基因定位概述
包括同源重组和非同源重组的遗传重组是生物演化的主要动力,也为基因定位提供了理论基础,从利用形态标记到目前大量使用的DNA分子标记进行基因定位,使定位结果的准确性和效率得到了大大提高,针对不同生物的遗传变异特点,产生了多种多样的基因定位方法,这些方法都是基于不同水平的遗传标记与性状的“连锁”和“关联”分析,即可根据它们之间的相关性程度定位基因的区域或位点(如SNP),然后通过定点突变等技术验证其功能(图1)。连锁定位主要考虑家系内个体之间的关系,通常先在连锁群上进行粗定位再精细定位,当基因标记与研究的基因相距越近时其定位越准确。关联定位利用性状与遗传变量(不同的基因标记)之间的统计相关性(P<0.01或P<0.05),关联定位比连锁分析更具统计把握度。

图1 基因定位
2 基因定位方法
2.1 连锁分析的基因定位
一般单基因遗传的质量性状多用连锁分析,在一个系谱/家系中,如果某一遗传标记/基因位点与某一性状存在“共分离”现象,则被认为它们有连锁证据,而位于同一染色体但距离较远或位于不同染色体上则认为控制性状的位点与基因标记位点不连锁。这种相关的模式在同一物种不同的群体中越一致,遗传标记/基因位点与性状的重组率越低,说明它们之间连锁强度越强,如果达到了“共分离”程度,这个标记位点可能是控制性状的基因位点,在后续的标记辅助育种选择、疾病基因检测等应用中越准确。
2.1.1 真核生物中的基因定位
真菌中的着丝粒作图[图2(a)]和植物中常用的三点测交进行的基因定位[图2(b)]都是利用了“连锁分析”的原理,着丝粒作图中把着丝粒看作一个基因位点,用着丝粒和其他基因的重组率来判断遗传模式和估算有连锁关系的着丝粒与基因之间的遗传距离。连锁定位的原理可以延伸至整个基因组上性状的基因定位。三点测交是经典的基因定位方法,如应用在果蝇、玉米等连锁群的绘制中。根据测交后代表现型的种类和比例判断基因间的遗传模式,并根据重组率估算连锁基因在染色体上的排列顺序和距离。

(a) 着丝粒作图;(b) “三点测交”进行基因定位。
2.1.2 细菌中的基因定位
细菌为单倍体,利用其同源重组进行基因定位有别于真核生物,其遗传重组包括结合、性导、转化和转导,表现出3个特点:(1) 遗传物质单方向转移;(2) 产生部分二倍体;(3) 基因只有整合到环状染色体上才能稳定遗传。利用结合、性导、转化和转导等遗传重组在细菌的基因定位中的应用如图3所示。
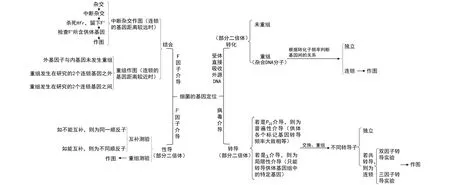
图3 细菌中的基因定位
2.2 关联分析的基因定位
多基因遗传的数量性状多用关联分析,关联研究指某一性状与某一等位基因之间的相关性,可以在候选基因定位、全基因组分析和连锁区域的精细定位上等进行有效分析,最早应用在人类的疾病基因定位中[16-17],后来在动植物性状的基因定位上应用较多[18-21]。本课题组用大豆的自然群体对菌核病抗性基因进行定位,定位到纤维素合成酶(CesA)、吡咯啉5羧酸还原酶(P5CR)等基因,并在甘蓝型油菜中证明了P5CR基因也与菌核病的抗性关联[22]。关联分析的基因定位通常有家系研究和病例-对照研究,前者研究的是有亲缘关系的个体之间等位基因频率,后者研究的是无亲缘关系的个体之间等位基因频率,但需要控制种族、性别和年龄等因素保持一致,即除了疾病以外的性状遗传背景尽量保持一致,在此情况下,等位基因频率在病例-对照组之间的差异可以解释为该等位基因与疾病/表型相关。
2.2.1 定位群体的选择
目前用来进行基因定位的群体有自然群体、分离群体(F2)、近等基因系(NILs)、重组自交系群体(RILs)、双单倍体群体(DH)、回交群体(BC)和多亲本高代互交系(MAGIC)等。不同群体的遗传背景各有特点,自然群体材料和F2群体材料创制容易,群体内遗传变异丰富,利于对多个不同性状同时进行关联定位,但群体一定要大,最好不少于200个样本,且用来定位的不同性状的表型变异要呈正态分布。NILs、RILs群体创制费时费力,除了特定性状的变异外其他性状的遗传背景一致,适合对特定性状的基因定位。如果某个性状的发育由多个基因调控,理论上其表型值呈现出一个范围较宽的连续变异。在遗传研究中,为了提高测序的性价比,可以采用极端表型进行基因定位[23-24],但要保证选择的定位群体是某个研究性状客观上的真正极端表型,所以尽量在大群体中选择极端类型,如图4所示,在小群体中选择的极端类型不能代表某一性状真正的极端表型,虽然也能定位出候选基因,但可能漏掉贡献率大的主效候选基因。
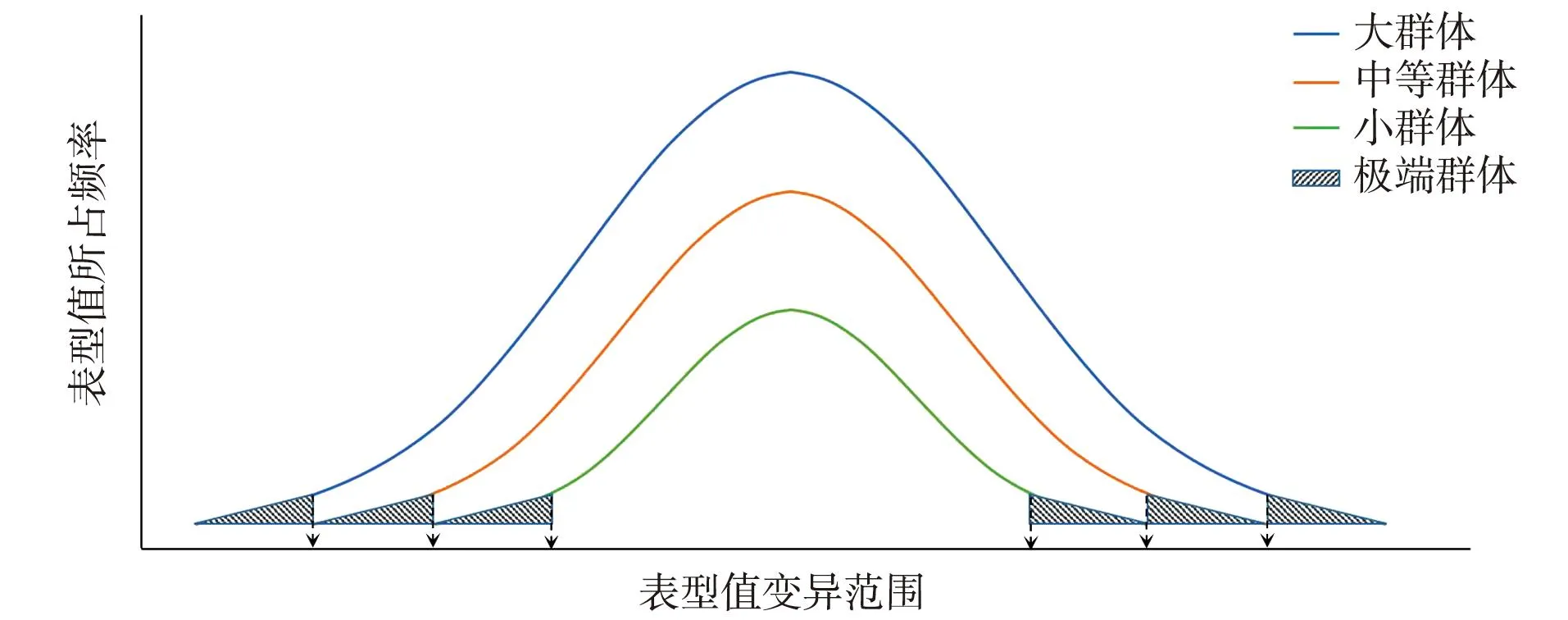
图4 大小不同群体的极端表型值分布
2.2.2 定位群体的基因型质量控制
在基因定位前利用如群体分层和Hardy-Weinberg平衡分析[25-26],对定位群体的基因型进行质量控制非常关键,可以降低结果的假阳/阴性的错误率。(1) 分层明显的群体不适合基因定位,因为群体分层导致基因定位产生很高的假阳性比率,通常用加大样本量、亲缘矩阵的前几个主成分进行矫正。(2) 偏离Hardy-Weinberg平衡有可能违反了一个或多个假设条件,是基因分型错误导致的。(3) 用动植物育种群体的遗传变异进行基因定位时不考虑Hardy-Weinberg平衡,因为这种群体有选择因素。(4) 如果是人类的病例群体,也不考虑Hardy-Weinberg平衡,因为偏离Hardy-Weinberg平衡的遗传变异位点可能与性状/疾病关联。(5) 由于遗传漂变使亚群内的遗传变异逐渐减少,亚群间的变异逐渐增多,因此,用具有遗传漂变的群体进行基因定位时,先进行莱特固定系数FST的估算。
2.2.3 关联分析定位基因流程
目前,根据全基因组测序结果产生的变异体主要包括SSR、SNP和InDel,其中,利用SNP及其构建的Haplotype进行基因定位是主要方向,由此产生的主流软件如PLINK、HaploView和TASSEL等都是用来进行全基因组关联基因定位研究的,其分析流程如图5所示。Haplotype往往是生物的大多数遗传变异根据遗传区段中的强连锁关系构成[27],Haplotype定位比SNP定位可提供更多的基因互作导致性状发育的证据[28],是目前对复杂性状进行基因定位最好的方法。
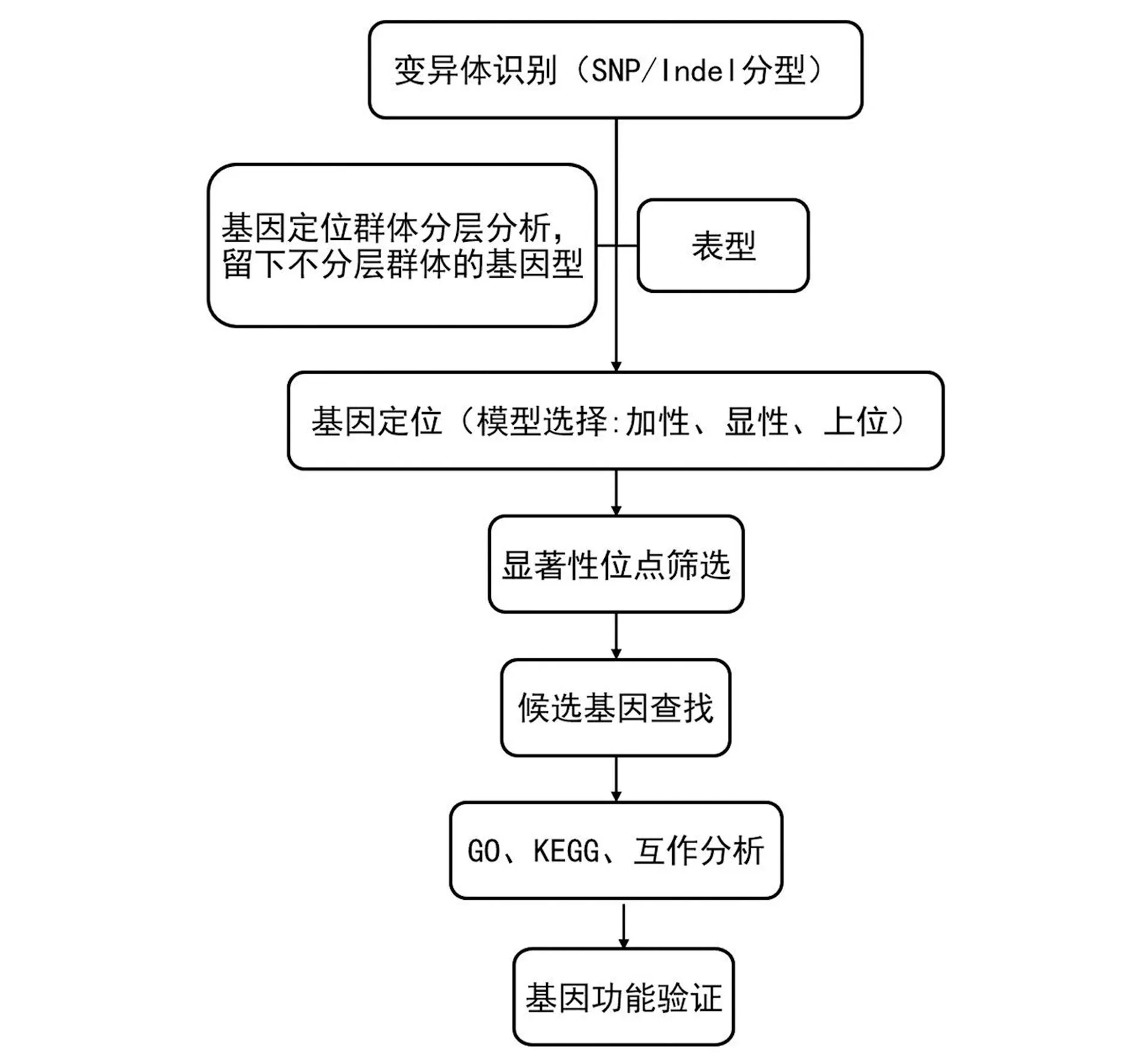
图5 全基因组关联研究中的基因定位分析流程图
3 结束语
遗传学作为生命科学领域里的引领学科,如何执行高校建设金课的“两性一度”(高阶性、创新性、挑战度)教学标准,取决于教师对新时代遗传学教学内容的把握深度和专业培养技能要求的理解。基因定位是各类遗传学课程研究的一项基本工作和重要环节,本文围绕基因定位所涉及的重点遗传学知识,以“连锁”和“关联”分析思路设计的几个相关教案,表现出“形散而神不散”的特点,能为学生的自主学习提供抓手,在教学过程中获得了较好的成效,同时为教师在遗传学教学过程中的教案设计提供参考。
猜你喜欢
新世纪智能(数学备考)(2021年9期)2021-11-24
现代装饰(2020年7期)2020-07-27
当代陕西(2019年15期)2019-09-02
福建基础教育研究(2019年10期)2019-05-28
NBA特刊(2018年7期)2018-06-08
现代装饰(2018年4期)2018-05-22
学苑创造·A版(2018年11期)2018-02-01
求学·理科版(2017年3期)2017-04-27
读者(2017年5期)2017-02-15
汽车维护与修理(2015年6期)2015-02-28
