
基于蒲地蓝药物组成碳点的制备及表征
2023-12-18 01:26吴晨涛阙子淼于诗源喻春皓
淮阴工学院学报 2023年5期
王 宇,吴晨涛,阙子淼,于诗源,喻春皓
(淮阴工学院 化学工程学院,江苏 淮安 223003)
近年来,由于碳点(carbon dots,CDs)光学性能优异[1]、生物相容性好[2]、低毒性等优点,许多专家学者开始对其开展研究。当前,制备CDs主要有自上而下和自下而上两种方法,将CDs从较大碳材料上剥离为自上而下,化学氧化[3]和激光腐蚀消融[4]都属于该方法,但此类方法需要较高的实验条件和特殊仪器;自下而上的方法则采用分子前驱体合成CDs,主要有微波辅助合成法[5]、超声合成法[6]、高温热解法[7]、水热法[8]等,这些方法大多使用化学试剂,容易污染环境,且表面改性等后续操作复杂,限制了其大规模生产和应用。因此,寻找廉价易得、天然无毒的碳源和简单便捷的制备方法是亟待解决的问题。与传统类型的CDs相比,生物质CDs具备更高的生物相容性、溶解性以及环境友好性。如藕粉[9]、茶叶[10]、蚕丝[11]等被作为碳源报道,以传统中药材为碳源,合成CDs却少有见闻。中药CDs具有碳源种类多、成本低、绿色环保、制备简单且可规模化生产等优势,熊威等[12]在2019 年从绵马贯众中制备出止血效果比较好的CDs;Wang等[13]将葛根作为原料合成葛根荧光碳点(PLR-CDs),研究表明该CDs 通过抑制XOD 活性,使尿酸降低。这些研究多以单味药材为碳源,对多种药材的组合CDs的制备基本没有报道。
蒲地蓝是具有清热解毒、消肿利咽等功效的中成药。蒲地蓝的清热解毒效果比蓝芩口服液更好,其抗炎消肿效果比二丁颗粒更好。蒲地蓝具有溶出速率快、药效稳定、药物毒性小及适用人群范围广等优点。该药以蒲公英作为君药,采用板蓝根为臣药加强君药作用,苦地丁、黄芩充当佐、使药解毒消肿。这些药材廉价易得,已有的研究对蒲地蓝的相关药理活性及质量检测也较为深入[14],因此,本文选取组成蒲地蓝的4 味中药为碳源,将其组合搭配制备CDs,这利于蒲地蓝药渣的再利用。另外,研究4味中药与其组合搭配的荧光强度的变化,可能是在反应过程中形成了某些新成分,为后续以中药为碳源进行深入研究提供了基础。
1 材料与方法
1.1 材料与试剂
黄芩、板蓝根、蒲公英、苦地丁均购于河北保定安国中药批发市场。硫酸奎宁(Quinine sulfate,QS)、硫酸(H2SO4)均购自阿拉丁中国上海。去离子水由学校提供。
1.2 实验仪器
电子分析天平(CP114);暗箱式紫外分析仪(郑州予泽仪器有限公司);电热恒温干燥箱(上海精宏实验设备有限公司);破壁粉碎机(倍艾家);医用离心机(湘仪离心机仪器有限公司);冷冻干燥箱(LGJ-10C,北京四环科学仪器有限公司);紫外可见近红外光谱仪(UV-2401,岛津);荧光分光光度计(F-7000,日本);傅立叶变换光度计(Nicolet 5700,美国);X射线衍射仪(D8Discover,德国)。
1.3 CDs的制备
对4 种中药碾磨成粉过40 目筛,中药按一定比例称取,中药总量为1.2 g。称取放置于100 mL聚四氟乙烯中,再量40 mL 去离子水倒入反应釜,超声约10 min后封装反应釜放置于电热鼓风干燥箱中。反应温度设置为180 ℃,反应时间为8 h。反应结束后离心30 min,收集上清液,取部分液体进行冷冻干燥,留待表征。具体的组分及比例搭配按表1进行配置。

表1 组成蒲地蓝的四味中药及组合搭配的荧光量子产率
1.4 CDs的量子产率计算
配制一定浓度的QS溶液,经适当稀释,控制紫外吸收光谱在360 nm 处吸光度低于0.05(AR),并记录此时的数据。将360 nm 作为激发波长,荧光发射光谱范围为370~620 nm,根据荧光图谱计算荧光积分峰面积(IR);CDs 溶液重复上述操作,吸光度和荧光积分峰面积分别记为A和I。本试验选取的QS 溶液是以0.1 mol∕L 硫酸溶液作为溶剂,对应的折射率(ηR)为1.33,CDs溶于去离子水中,其η值为1.33[11]。
1.5 CDs的表征
采用紫外-可见分光光度计和荧光光谱仪对4味中药及其组合搭配的15种CDs进行光学性能的检测。选取一类搭配下荧光效果最好的一组,进行激发光谱和荧光光谱的测量,同时也考察了不同激发波长的影响。设置激发波长为360 nm,狭缝宽度为5 nm,扫描速度为2 400 nm∕min,光电倍增管电压为500 V。后续用傅立叶红外光谱仪(FT-IR)、X 射线衍射仪(XRD)对选取的CDs 进行形貌、官能团的表征。
2 结果与讨论
2.1 CDs光学性能分析
图1(a)中4种中药制备的单一CDs,在283 nm处都存在最大紫外吸收峰,在稀释相同倍数下,板蓝根CDs(CD3)吸光度明显优于其他CDs。图1(b)为该浓度下CDs 的荧光光谱检测,发现CD3的荧光效果最好,其次为蒲公英CDs(CD2),黄芩CDs(CD4)最差。在360 nm 激发波长下,4 种CDs 的发射峰均为425 nm。
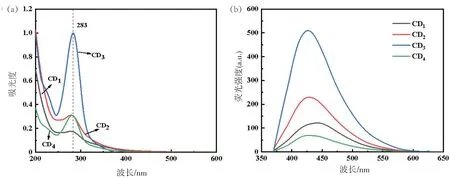
图1 单一中药CDs紫外光谱图(a)和荧光光谱图(b)
图2(a)为中药两两组合下共6 种CDs,与单一CDs相同,最大吸收峰都处于在283 nm处,与图1(a)相比,经配伍后的两两CDs 吸光度差异不大。如图2(b)所示,对CDs 进行荧光强度的测量,与单一中药相比,荧光强度较为集中且分布相对均匀,比较6 种CDs 的荧光强度发现板蓝根-蒲公英CDs(CD8)性能最优越,与图1(a)中单一CDs荧光强度顺序呈较好搭配。

图2 两种中药复合的CDs紫外光谱图(a)和荧光光谱图(b)
图3(a)为中药三三组合下的4 种CDs,其最大吸收峰仍为283 nm;图3(b)的荧光光谱显示,板蓝根-蒲公英-苦地丁CDs(CD11)荧光强度优于其他同种配伍的CDs,与单一CDs与两两CDs的最佳荧光强度顺序相符合。
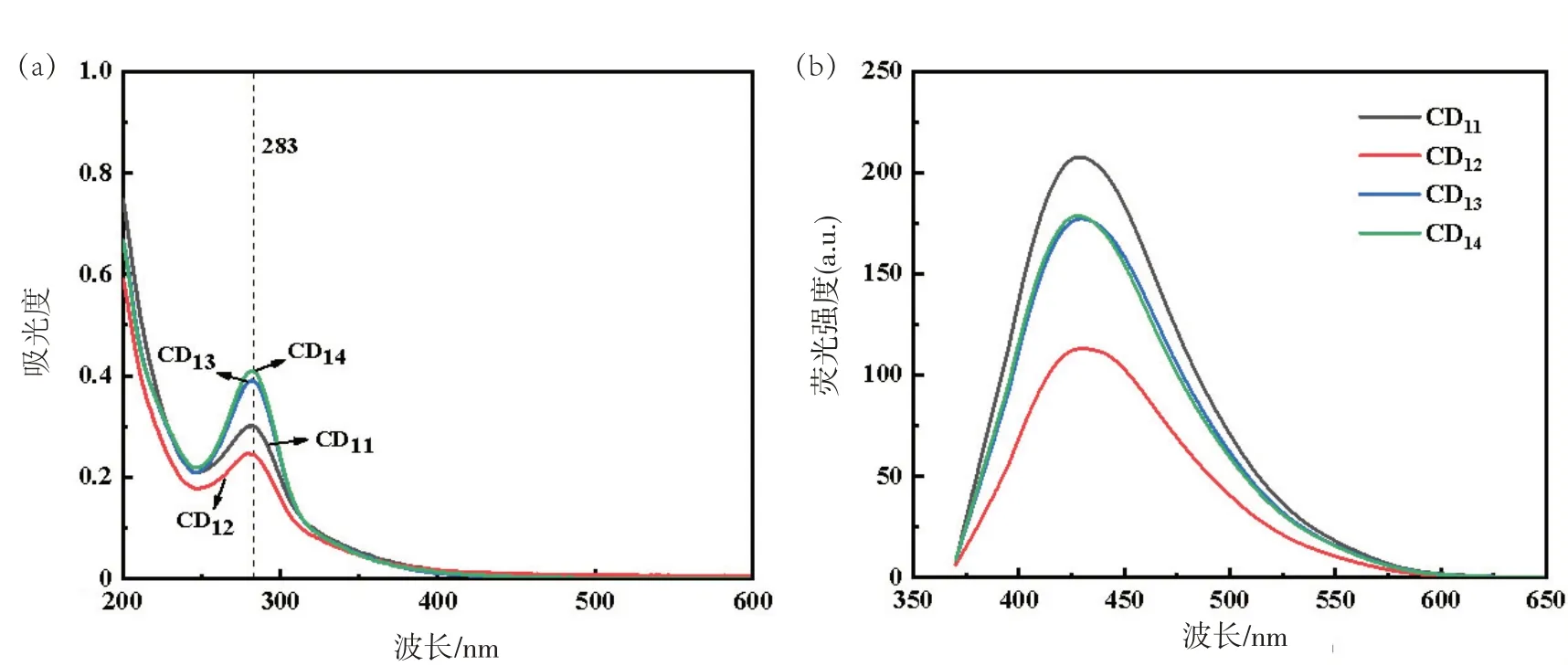
图3 三种中药复合的CDs紫外光谱图(a)和荧光光谱图(b)
图4(a)4 种中药的混合搭配为蒲地蓝CDs(CD15),与上述组合相同。其最大吸收峰仍为283 nm;图4(b)中CD15的荧光强度与图2(b)、图3(b)的复合CDs无明显差异。
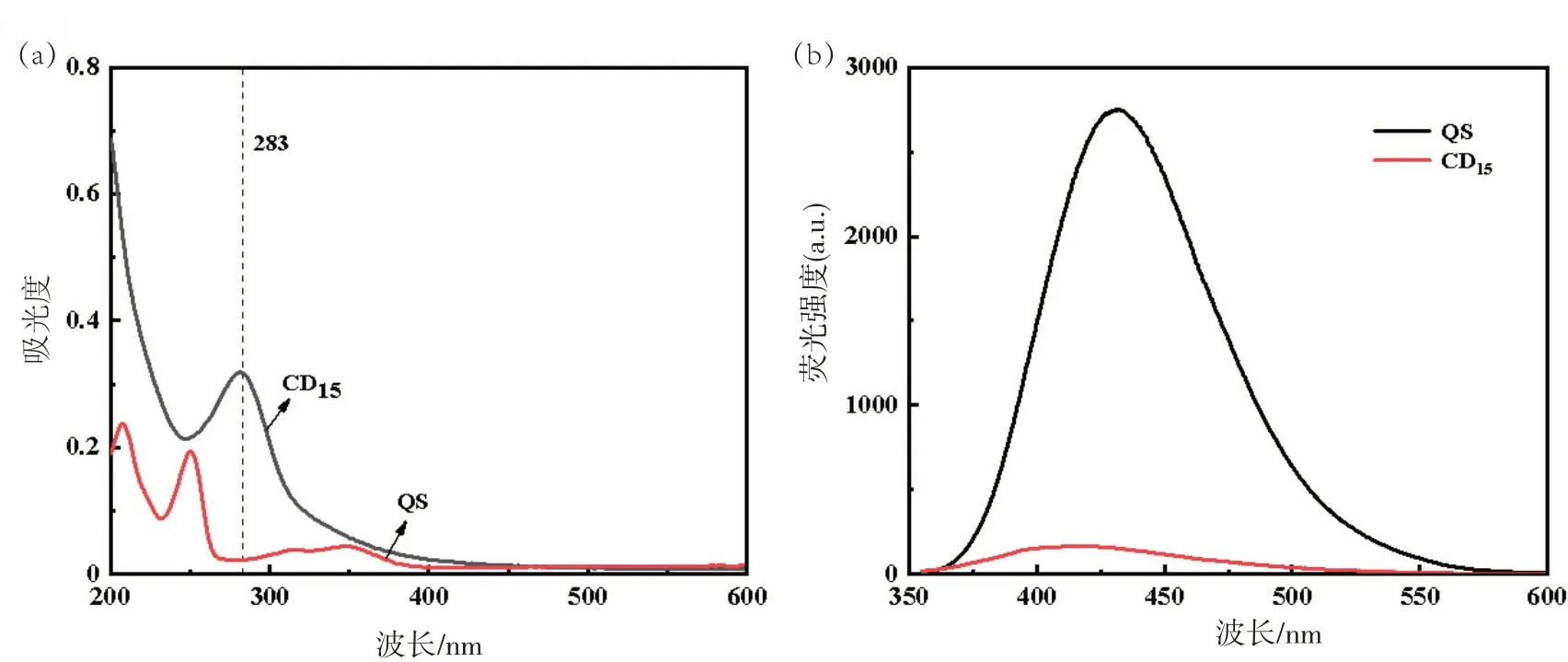
图4 四种中药复合的CDs及QS紫外光谱图(a)和荧光光谱图(b)
图1~图4 的紫外图谱显示,15 种CDs 均在283 nm处存在最大吸收峰,可能是sp2杂化C=C键的π-π*跃迁。可见光区域吸收较弱,无明显特征峰。QS 在220,260,360 nm 处有明显吸收峰。由于两者都在360 nm 处存在较强吸收,后续进行量子产率计算时以360 nm作为激发波长。
在360 nm 波长激发下,比较15 种CDs 的荧光光谱,CD3的荧光强度最强,可能是板蓝根内含有靛蓝等成分,存在较大的共轭体系,所以荧光性能较为优越。在后续搭配中,每类组合下荧光强度最好的一个,符合单一CDs荧光强度的顺序,且15种CDs在360 nm激发波长下发射峰均于425 nm附近。后续将QS 作为参比液,计算荧光积分峰面积得到碳量子产率。为进一步探讨CDs的光学性能,选取每类荧光效果最好的,依次为CD3、CD8、CD11、CD15进行了激发光谱和发射光谱的测量,同时比较不同激发波长对CDs荧光强度的影响。
图5(a)CD3的最佳激发波长为325 nm,最佳发射波长为400 nm;图5(b)中,将CD3的激发波长从320 nm 以20 nm 为增量增加至400 nm,其发射峰的位置由400 nm 偏移到463 nm,且荧光强度下降。
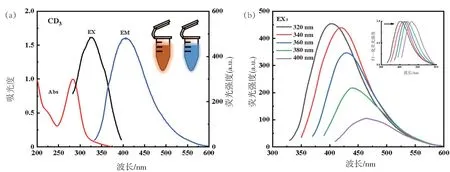
图5 CD3的激发和发射光谱图(a)和在不同激发波长下荧光光谱及归一化谱图(b)
图6(a)CD8的最佳激发波长为300 nm,最佳发射波长为390 nm;图6(b)中,CD8与CD3测量条件相同,在荧光强度依次下降的情况下,发射峰的位置从390 nm偏移到463 nm。
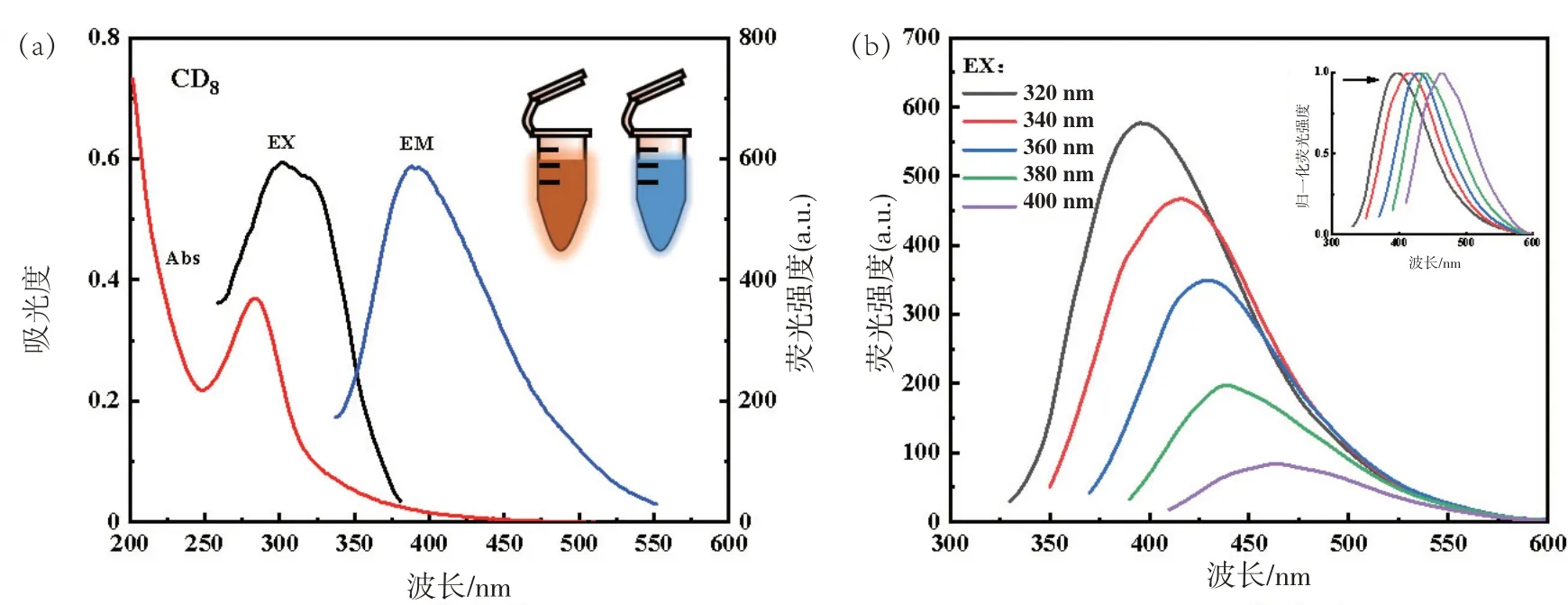
图6 CD8的激发和发射光谱图(a)和在不同激发波长下荧光光谱及归一化谱图(b)
图7(a)CD11的最佳激发波长为300 nm,最佳发射波长为390 nm;如图7(b),CD11与CD8相似,在改变激发波长时,荧光强度发生明显变化,且发射峰的位置从395 nm处向463 nm偏移。
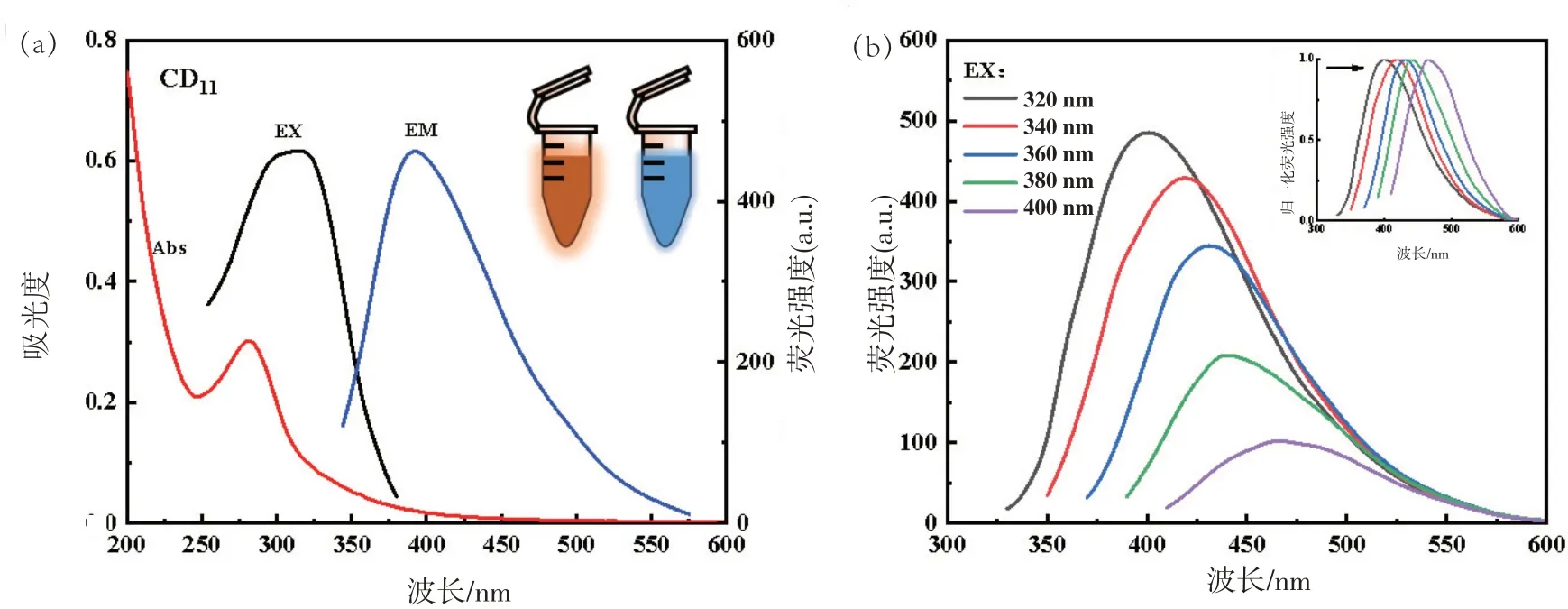
图7 CD11的激发和发射光谱图(a)和在不同激发波长下荧光光谱及归一化谱图(b)
图8(a)CD15最佳激发波长为300 nm,最佳发射波长为390 nm;图8(b)显示,CD15的荧光强度随激发波长的增大而下降,发射峰的位置由395 nm向464 nm偏移。
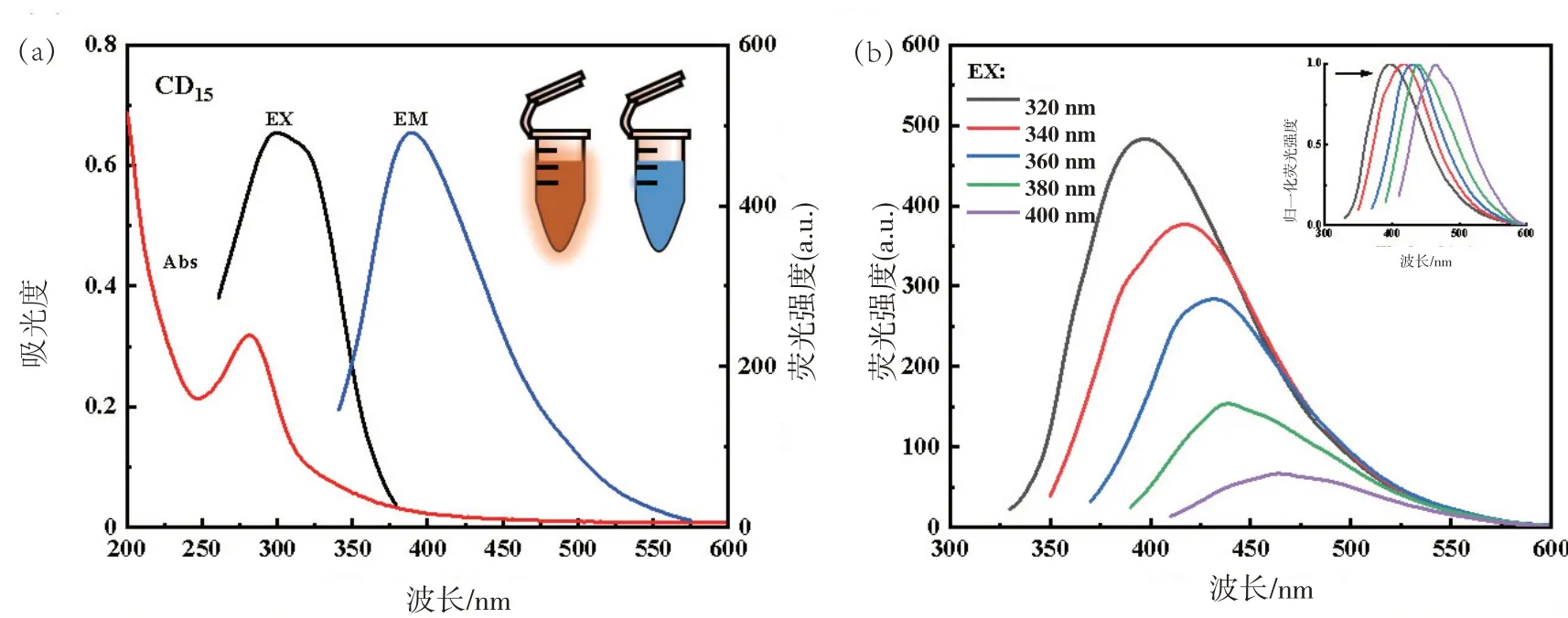
图8 CD15的激发和发射光谱图(a)和在不同激发波长下荧光光谱及归一化谱图(b)
CDs 溶液在正常光源照射下为淡黄色,365 nm紫外灯下为蓝色荧光。图5(a)中CD3在最佳激发波长为325 nm,最佳发射波长为400 nm,随着对中药进行组合,图6(a)中的CD8、图7(a)中的CD11、图8(a)中的CD15最佳激发波长和发射波长开始向短波长方向偏移,最佳激发波长为300 nm,最佳发射波长位于400 nm处。可能是中药组合后CDs内部发生变化引起的。后续,探讨不同激发波长下CDs的荧光图谱。分析得到,4种CDs在激发波长320~400 nm范围内荧光峰位置发生变化。通过对不同激发波长做的归一化图谱可以看出红移现象十分明显。表明制备的CDs具有激发依赖性。
2.2 CDs量子产率分析
表1 中列出组成蒲地蓝的4 味中药及11 种组合搭配的碳量子产率,根据中药组合的个数,单一中药制备的CDs,量子产率最高的为CD3,达到17.80%;两两组合的CDs,CD8效果最好;三三搭配的CDs,CD11产率更高;CD15为蒲地蓝CDs,其量子产率为2.54%,通过比较,发现CDs 的量子产率与测得的荧光强度基本上呈正比。CD3的量子产率远比其他CDs呈现出更高的量子产率,可能是板蓝根中含有靛蓝等物质,其荧光发射能力更大,因此量子产率更佳。经组合后的CDs 量子产率相对较低,可能是由于靛蓝与其他中药成分发生干扰,从而荧光强度衰弱。
2.3 CDs的FT-IR图谱分析
图9 为CD3、CD8、CD11、CD15的FT-IR 图谱 显示,在3 000~3 750 cm-1波数范围内均存在一个宽而大的吸收,该吸收属于N-H 和O-H 的伸缩振动吸收区域,在1 602 cm-1和1 400 cm-1的吸收归属于C=O、C=C 伸缩振动。1 092 cm-1为C-O 的弯曲振动吸收峰。根据上述分析可以得到CDs 表面含有羟基,氨基等官能团。
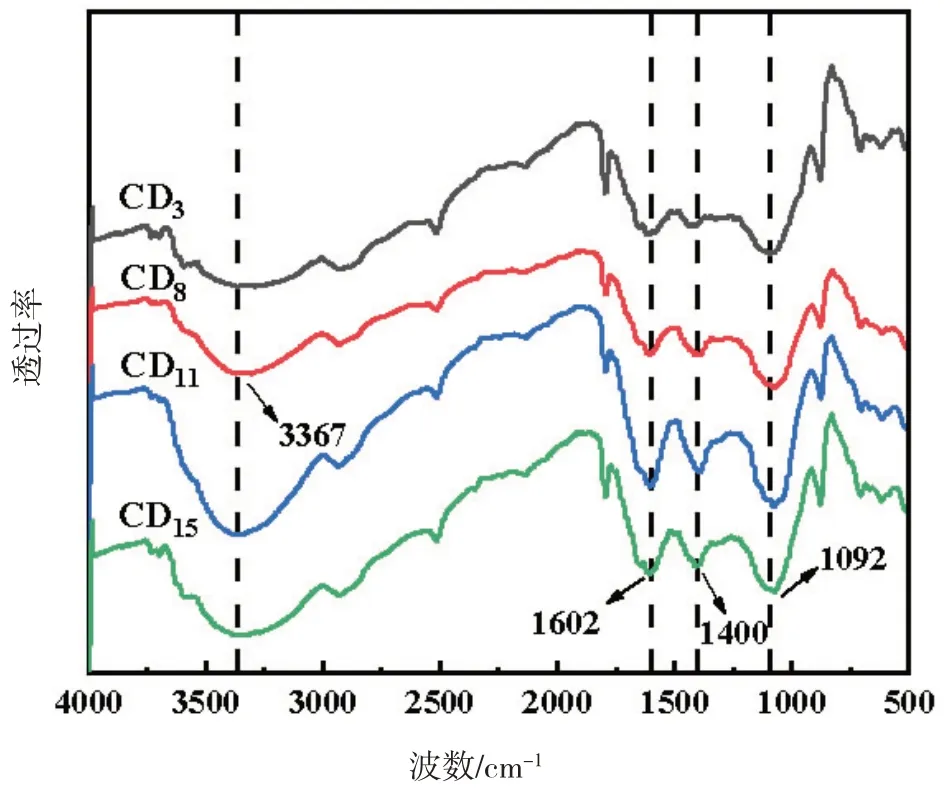
图9 CDs的FT-IR光谱图
2.4 CDs的XRD图谱分析
如图10 所示,CD3的XRD 图谱显示出一个很宽的峰,峰中心在21.62°,说明该CDs 晶格存在缺陷,属于无定形碳。当中药类别增加,CD8、CD11、CD15的XRD 图谱峰的位置发生改变,依次变为22.11°、23.89°、24.07°,可能是中药组合后CDs内部发生了某些新的作用。
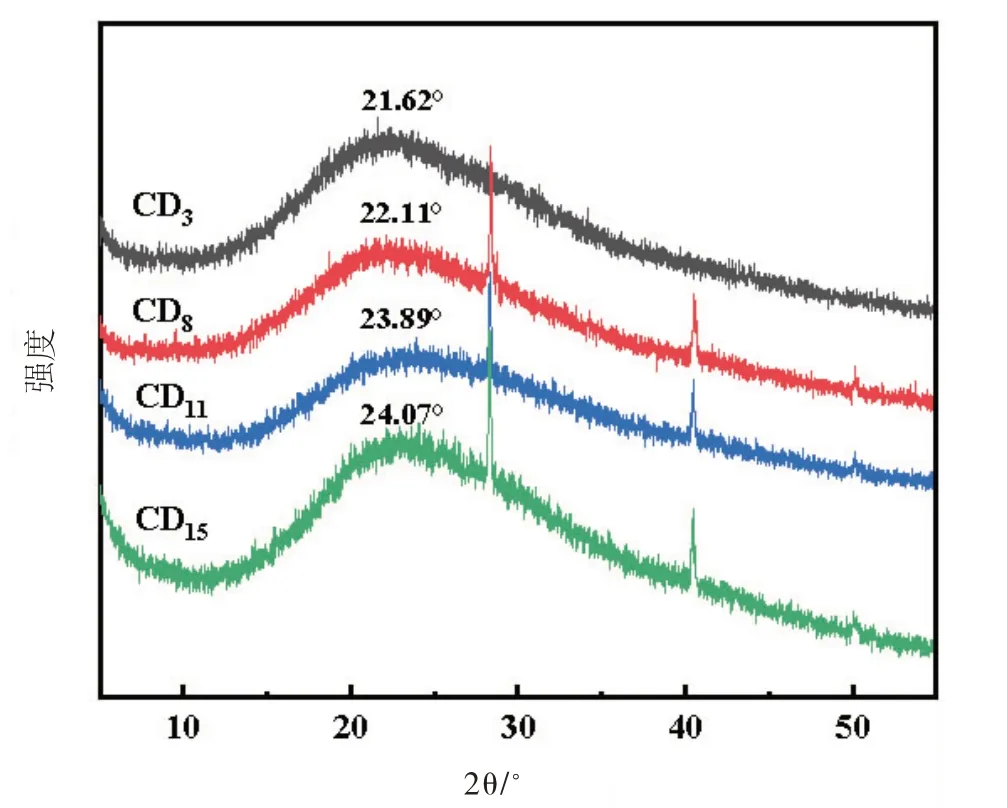
图10 CDs的XRD图像
3 结论
本文采用组成蒲地蓝的4 味中药及其组合搭配为碳源,通过水热法制备15 种CDs。以荧光强度为条件,筛选出每类搭配中效果最好的CDs,进行激发、发射光谱的检测,同时考察不同激发态下发射光谱的变化。用FT-IR、XRD对4种CDs进行表征,分析CDs 的表面结构。结果表明,CDs 在365 nm 紫外灯下呈蓝色荧光,于283 nm 处存在最大紫外吸收峰,激发波长360 nm 荧光光谱中发射峰位均在425 nm。与CD3相比,CD8、CD11、CD15的激发光谱和发射光谱发生蓝移,激发波长由325 nm向300 nm移动,发射光谱由400 nm向短波长偏移,可能是由于药材组合使得CDs内部发生变化。根据FT-IR 图谱及XRD 图谱分析,CDs 表面含有羟基,氨基以及羧基等官能团。中药CDs制备主要是单味药材,混合中药制备CDs基本没有报道,本文为中药CDs的制备提供新的思路,后续应用中,提高CDs的量子产率也是需要解决的重点问题。
猜你喜欢
实验室研究与探索(2022年8期)2022-11-12
中老年保健(2022年4期)2022-08-22
世界科学技术-中医药现代化(2021年9期)2021-12-31
中国测试(2021年10期)2021-11-12
中国民间疗法(2020年22期)2021-01-07
光学仪器(2020年3期)2020-07-10
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
益寿宝典(2018年35期)2018-01-26
中成药(2017年12期)2018-01-19
