
蒙东地区三种药用植物根际微生物群落特性研究
2023-12-18 11:47寇佩雯刘长乐许祎珂宋忠兴李铂张永生黄文静唐志书
中国农学通报 2023年33期
寇佩雯,刘长乐,许祎珂,宋忠兴,李铂,张永生,黄文静,唐志书,3
(1陕西中医药大学/陕西中药资源产业化省部共建协同创新中心/秦药特色资源研究开发国家重点实验室(培育),陕西咸阳 712083;2内蒙古海天制药有限公司,内蒙通辽 028000;3中国中医科学院,北京 100700)
0 引言
在《本草经集注》中有“诸药所生,皆有境界”的记载,同样在实际生产中也发现环境改变可以引起中药质量的改变[1]。研究发现,除生态环境和逆境胁迫等影响药用植物道地性的因素外,药用植物根际微生物亦是影响药材道地性的因素之一[2]。全而平衡的土壤微生物对于保证药用植物正常生长、发育、和代谢具有重要意义[3]。1904年德国微生物学家Lorenz Hiltner提出“根际”概念时指出,根际微生物在一定程度上可以抑制经土传染的植物病害,植物根际微生物参与了土壤中许多养分的转化,直接或间接地影响了自然生态系统中植物群落的组成、生物量和功能[4]。土壤微生物在一定程度上影响了植物的产量和植物的修复效率[5]。微生物活性可以改变根系渗出物的组成[6-7]。研究发现,根际微生物可以通过影响药用植物的抗病虫害能力、抗逆能力、药用部位的生长情况、有效成分的积累和连作障碍等来影响药用植物的品质[8-9]。与此同时,植物也会根据环境变化释放不同的根系分泌物影响微生物的结构以适应周围环境[10]。植物对微生物的影响可以通过改变渗出物中碳水化合物、羧酸和氨基酸的含量,从而影响植物的根际环境。研究发现,当根系环境中所含铁和磷的可用性不足时,植物会渗出有机酸阴离子来刺激根际微生物酸化土壤,从而改善自身生存环境并获取营养物质[11]。
根际微生物群落特征是由农业管理和宿主选择过程之间的相互作用所决定的[12]。在拟南芥等植物的研究中发现,不同植物种类、同一植物的不同基因型和植物的不同生长发育时期都对根际微生物的组成有明显影响[13-15]。土壤的物理化学性质、肥力、pH等因素可以通过影响植物的生理特性和根际沉积的组成进一步影响植物根际微生物群落的结构[16]。对沙草的研究发现,土壤的pH等性质会影响沙草根际微生物群落的结构[17]。在4 种草莓栽培品菌根真菌的研究中发现,土壤的物理化学特性和管理措施决定了特定宿主相关的微生物群落特性和结构[18]。可见,根际微域内的土壤、土壤微生物和植物根系间相互作用相互影响,不同药用植物的根际微生物具有独特的特征。药用植物根茎叶中的各类代谢物或次生代谢物是中药药理的主要成分,植物的根际和叶际微生物可能对这些代谢物的产生有一定的促进或抑制作用,同时这些物质也可能对微生物群落产生影响。因此,探究药用植物根际微生物群落的特征,以及土壤环境因子和植物类型对微生物群落的影响情况对指导药用植物种植,保证药用植物质量具有重要意义[19]。
内蒙古是中国药用资源大省,拥有丰富的药材品种和广阔的种植面积[20]。该地区种植有黄芪、黄芩、桔梗、麻黄、防风、北沙参、甘草等多种中药材[21]。研究发现,甘草、防风和北沙参在内蒙古地区种植面积较广[22-24]。本研究采集了内蒙古蒙东地区种植年限、长势相近的甘草(GlycyrrhizauralensisFisch.)、北沙参(GlehniaeRadix)和防风(Saposhnikoviadivaricata)三种药用植物的根际土壤以及实验基地的非根基土壤为样本,测定了各样本的土壤理化性质,并通过高通量测序技术对各样本的微生物进行了初步研究,旨在分析内蒙地区小的局域环境中药用植物根际和非根际土壤中微生物的结构和丰度特征,揭示土壤环境因子和植物类型对微生物群落的驱动效应。
1 材料与方法
1.1 研究区域和采样点的设置
在内蒙古通辽市科尔沁区庆和镇林场(经纬度:121°55′17′′E,43°43′56′′N;海拔:140 m)3号地试验田,于2021年9月11日采用多点混合法进行取样,各样本各选取3个采样点。对照组(CK)样本选取自距离实验田50 m左右未种植过植物的非根际土壤,根际土壤采集自长势和株龄(约1 年)相近的防风(FF)、甘草(GC)和北沙参(SS)。非根际土壤采集时先去除地表5 cm厚的土壤和覆盖物,然后收集0~20 cm 深度的土壤。根际土壤收集采用抖落法,即在每个采样点选取5 株植物,收集附着于植株根系表面约0.5 cm 左右的土壤。随后,将相同样品混匀后分为两份,分别装于无菌密封袋中,封存于干冰盒中,并送至上海派森诺生物科技有限公司进行土壤理化性质分析和微生物群落分析。
1.2 土壤理化性质测定
土壤理化性质测定主要参考《土壤农化分析》[16]进行,采用浸提法利用ORP-3000酸度计测定pH;采用采用浓硫酸消解-凯式定氮法测定全氮(TN)含量;采用钼锑显色法测定全磷(TP)含量;采用火焰光度法测定全钾(TK)含量;采用化学比色法测定脲酶(URE)、蔗糖酶(SUC)、过氧化氢酶(CAT)和酸性磷酸酶(AP)的含量。
1.3 土壤微生物群落分析
利用FastDNA®Spin Kit for Soil试剂盒提取各样本土壤的总DNA,用NanoDrop ND-2000对DNA进行定量,后通过1.2%琼脂糖凝胶电泳检测DNA 质量。采用全式金公司的Pfu 高保真DNA 聚合酶进行PCR扩增,利用338F(5’-ACTCCTACGGGAGGCAGCAG-3’)和806R(5’-GGACTACHVGGGTWTCTAAT-3’)引物对细菌V3-V4 区进行扩增;利用SSU0817F(5’-TTAGCATGGAATAATRRAATAGGA-3’)和1196R(5’-TCTGGACCTGGTGAGTTTCC-3’)引物对真菌V5-V7区进行扩增。扩增产物经磁珠纯化回收,后采用Quant-iT Pico Green dsDNAAssay Kit进行荧光定量检测,根据荧光定量结果,按照每个样本的测序量需求对各样本按相应比例等摩尔混合。混合后的扩增产物以Illumina 公司的TruSeq Nano DNA LT Library Prep Kit制备测序文库。
对制备好的文库使用Agilent Bioanalyzer 进行质检,合格的文库采用Illumina MiSeq 测序仪进行双端测序,原始下机数据根据序列质量进行初步筛查,并根据index 和Barcode 信息对文库和样本进行划分。按照QIIME2 dada2分析流程和Vsearch软件的分析流程进行序列去噪和操作分类单元(OTUs)聚类,对比Silva数据库(Release132,http://www.arb-silva.de)进行物种分类学注释,并采用RDP classifier 贝叶斯算法对在97%相似水平下的OTU进行丰度分析,揭示样品的物种组成。根据OTU在不同样本中的分布,评估每个样本的α 多样性水平,包括Coverage、Chaol、ACE 和Shannon等指数分析。
1.4 数据统计与分析
使用SPSS 26软件对样品土壤理化变量数据进行单因素方差分析(ANOVA),并采用新复极差法(Duncan)比较数据组间差异。使用Pearson 相关系数对各个指标进行相关性分析。使用R语言包vegan进行群落α 多样性分析,并利用Microsoft Excel 2013 对数据进行整理和制表。通过派诺森基因云平台(https://www.genescloud.cn/home)绘制相关性分析热图,通过图图云平台(https://www.cloudtutu.com/#/login)绘制垂直分组柱状图、RDA冗余分析图和方差分解分析图。
2 结果与分析
2.1 土壤理化性质分析
由表1可知,各样本之间的TN、TK、URE、SUC和AP 存在显著差异。其中,TN、URE、SUC 和AP 均在GC中最大,在CK中最小。TK在CK中最大,在SS中最小。各样本间pH没有显著差异,但所有土壤样本的pH都大于8,为弱碱性土壤。TP和CAT仅在非根际土壤(CK)和根际土壤(FF、GC 和SS)之间存在显著差异(P<0.05),TP 含量在非根际土壤中较高,而CAT 含量在非根际土壤中较低。可见,不同样本之间的土壤理化性质存在较大差异,尤其是在根际土壤与非根际土壤之间。
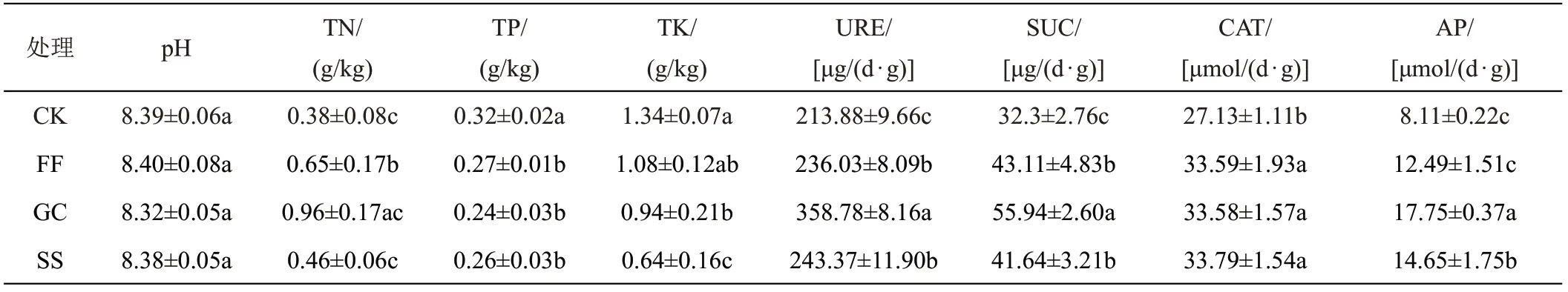
表1 各土壤的理化性质
2.2 非根际及3种药材根际微生物群落组成及多样性
在97%的相似度水平下,对土壤样品中的细菌进行了OTU 个数的鉴定,并对OTU 代表序列进行物种注释。在土壤样本中共检测出细菌有36 个门、105 个纲、231个目、402个科、770个属和1594个种,在3种植物的根际土壤中共检测出细菌38 个门、110 个纲、244个目、443个科、966个属和2272个种。在土壤样本中共检测出真菌12个门、30个纲、59个目、126个科、210个属和269 个种,在3 种植物根际中共检测出真菌14个门、39 个纲、86 个目、186 个科、327 个属和495 个种。可见在该微域环境的根际土壤中微生物丰富且多样性较高。
在所有4组样本中,前15个优势细菌门分布情况如图1A 所示,其中,丰度大于1%的菌群,CK 中有8个,根际土中SS、FF、GC 分别各有8 个、7 个和6 个。CK、FF和SS的优势菌门分布相似,前3个优势菌门分别为变形菌门 (Proteobacteria)、放线菌门(Actinobacteria)和酸酐菌门(Acidobacteria)。而GC 与其他样本不同,前3个优势菌门分别为变形菌门、放线菌门和拟杆菌门(Bacteroidetes)。如表2 所示,在所有样本中,GC 细菌群落的Shannon 指数和Simpson 指数最低,FF细菌群落Shannon指数最高,CK和SS细菌群落的Simpson 指数相同且最高。可见GC 中细菌群落多样性最低,均匀度也较低,FF 中细菌群落多样性最高。CK和SS中细菌群落均匀度最高。FF的Chao1指数和ACE指数最高,可见其根际细菌群落中种类数目最多,而GC样本与FF样本相反。
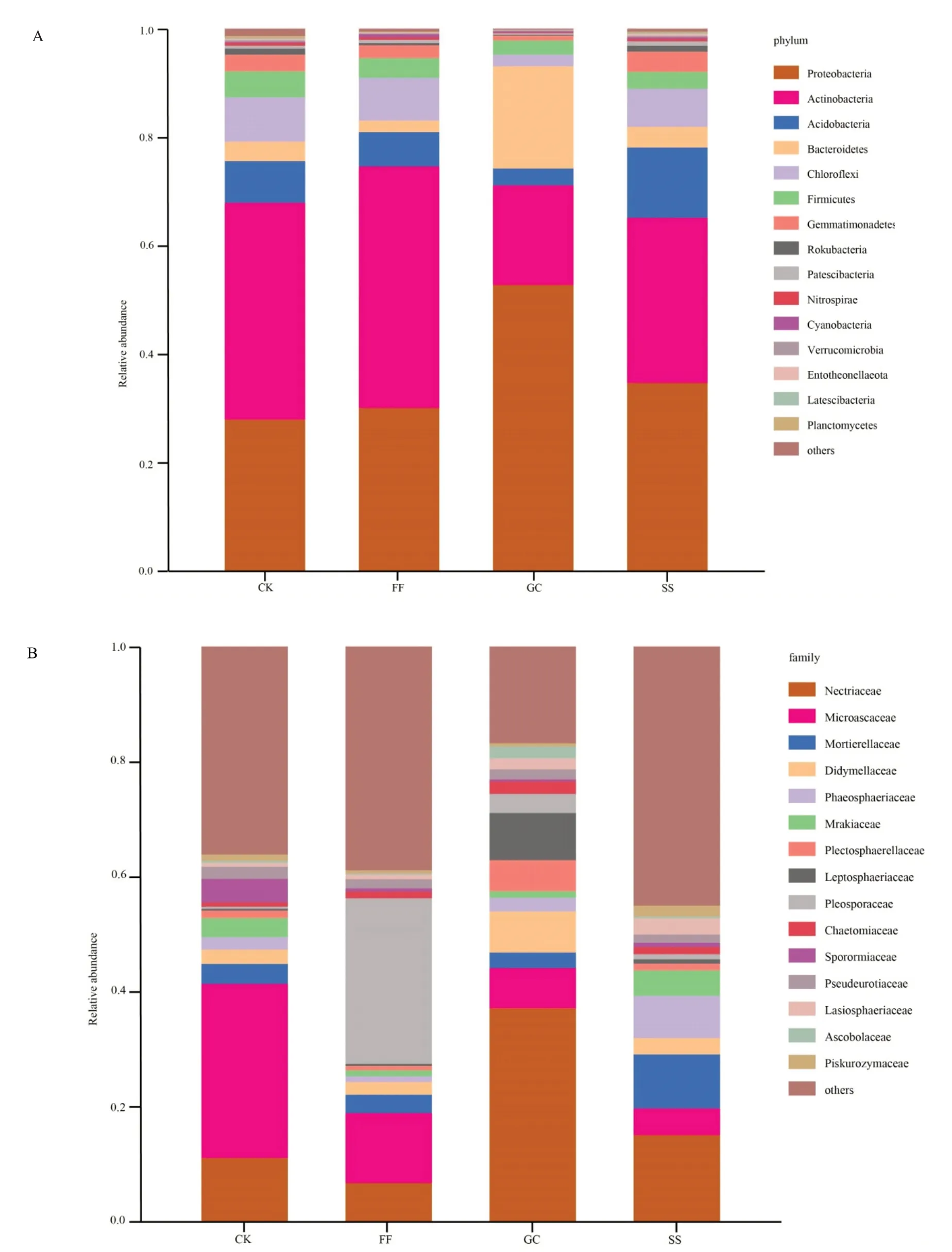
图1 不同样本细菌优势菌门(A)和真菌优势菌科(B)的相对丰度
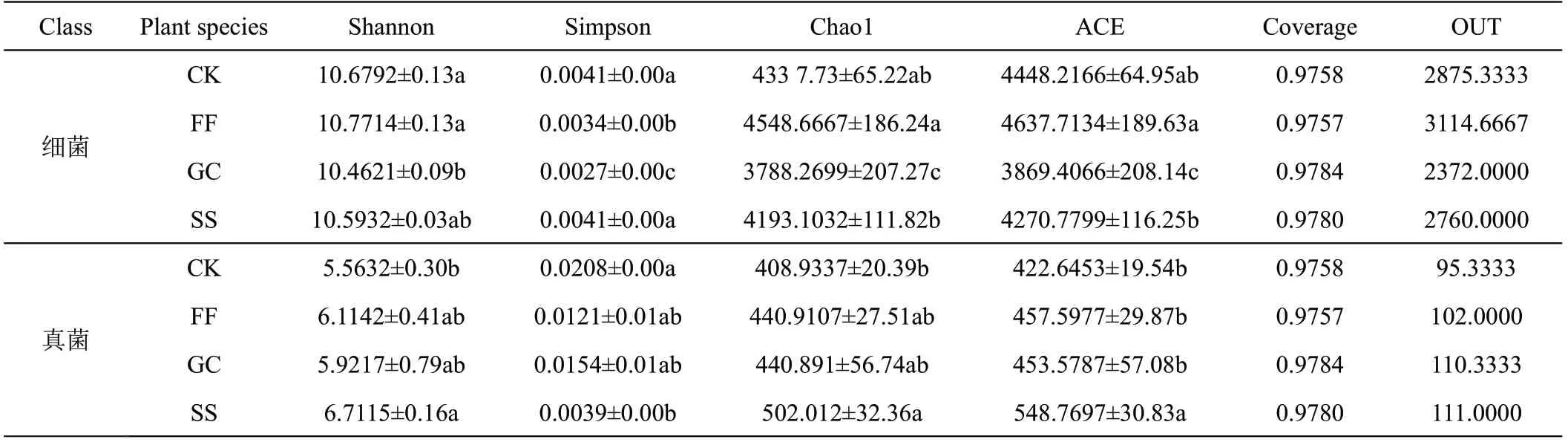
表2 非根际及3种药材根际土壤细菌和真菌群落多样性
所有4 组样本中,前15 个优势真菌科组成,如图2B 所示,其中丰度大于1%的菌群,在CK 中有10 个,根际土中GC、SS、FF 中分别有13 个、11 个和9 个。各样本中排在前列的优势真菌科分布不完全相同,除赤壳科(Nectriaceae)和微囊科(Microascaceae)均位于各样本优势菌科前列外,FF 中孢子科(Pleosporaceae)、GC中鳞毛蕨科(Phaeosphaeriaceae) 和被孢霉科(Mortierellaceae)、SS 中二甲藻(Didymellaceae)和球菌科(Leptosphaeriaceae)也为各样本中占比较大的菌科。如表2所示,SS真菌群落的Shannon指数最大,而Simpson 指数最小。CK 真菌群落的Shannon 指数最小,可见CK真菌群落多样性较小,SS真菌群落多样性最高而均匀度最低。GC的Chao1指数和ACE指数最低,由此说明其根际真菌群落中种类数目最少,而SS的2个指数和根际真菌群落中种类数目与GC相反。
2.3 环境因子对土壤微生物群落的影响
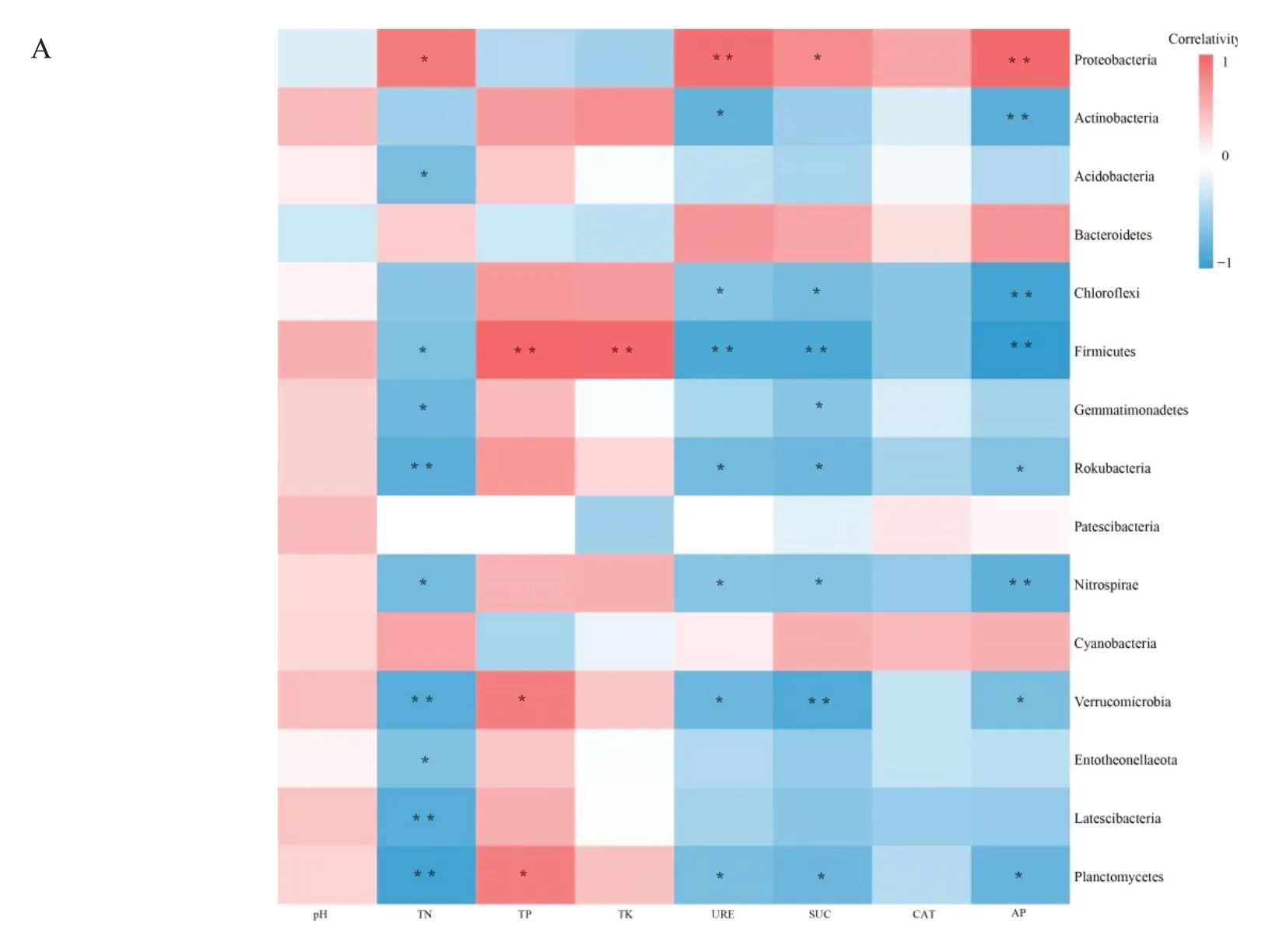
不同的环境因子对微生物群落结构多样性具有显著性影响。如表3 所示,利用Spearman 方法分析土壤环境因子与非根际和根际中菌群α多样性指数间相关性。发现对于细菌菌群,Shannon指数和Chao1指数与AP 负相关,Simpson 指数与TN、URE 和SUC 负相关,ACE 指数与URE 负相关。而对于真菌群落CAT 与Shannon 指数、ACE 指数和Chao1 指数正相关,与Simpson 指数负相关。TK 与Simpson 指数正相关,与Shannon 指数、ACE 指数和Chao1 指数负相关。可见在该局域环境内全氮、脲酶、蔗糖酶和酸性磷酸酶对细菌群落结构影响较为显著,全钾和过氧化氢酶对真菌群落结构影响较显著。
运用Spearman 分析筛选土壤理化性质中各菌群群落构成的驱动因子。由图2A 和2B 可知,土壤环境因子主要通过影响变形菌门、厚壁菌门、疣微菌门和浮霉门影响了细菌菌群分布,又通过影响丛赤壳科、微囊科、假球壳科和球菌科影响了真菌菌群结构的分布。
为了探究驱动各菌群差异的环境因素,对各样本的菌群丰度和显著相关的5项土壤理化因子进行冗余分析。由图3A和3B可见,在细菌菌群和真菌群落中,除FF菌群分布与CK较为接近外,其余各菌群分布差异明显,并且GC 菌群分布较其他群落较远。分析群落组成结构与环境因子的关系。可见在细菌门水平上第一轴和第二轴的解释率分别为66.21%和8.04%,总解释率达74.25%。在真菌科水平上,第一轴和第二轴的解释率分别为34.83%和16.59%,总解释率达51.42%。综合来看,样本的菌群结构在细菌门水平上和真菌科水平上受TN、TP、URE、AP和SUC的影响。
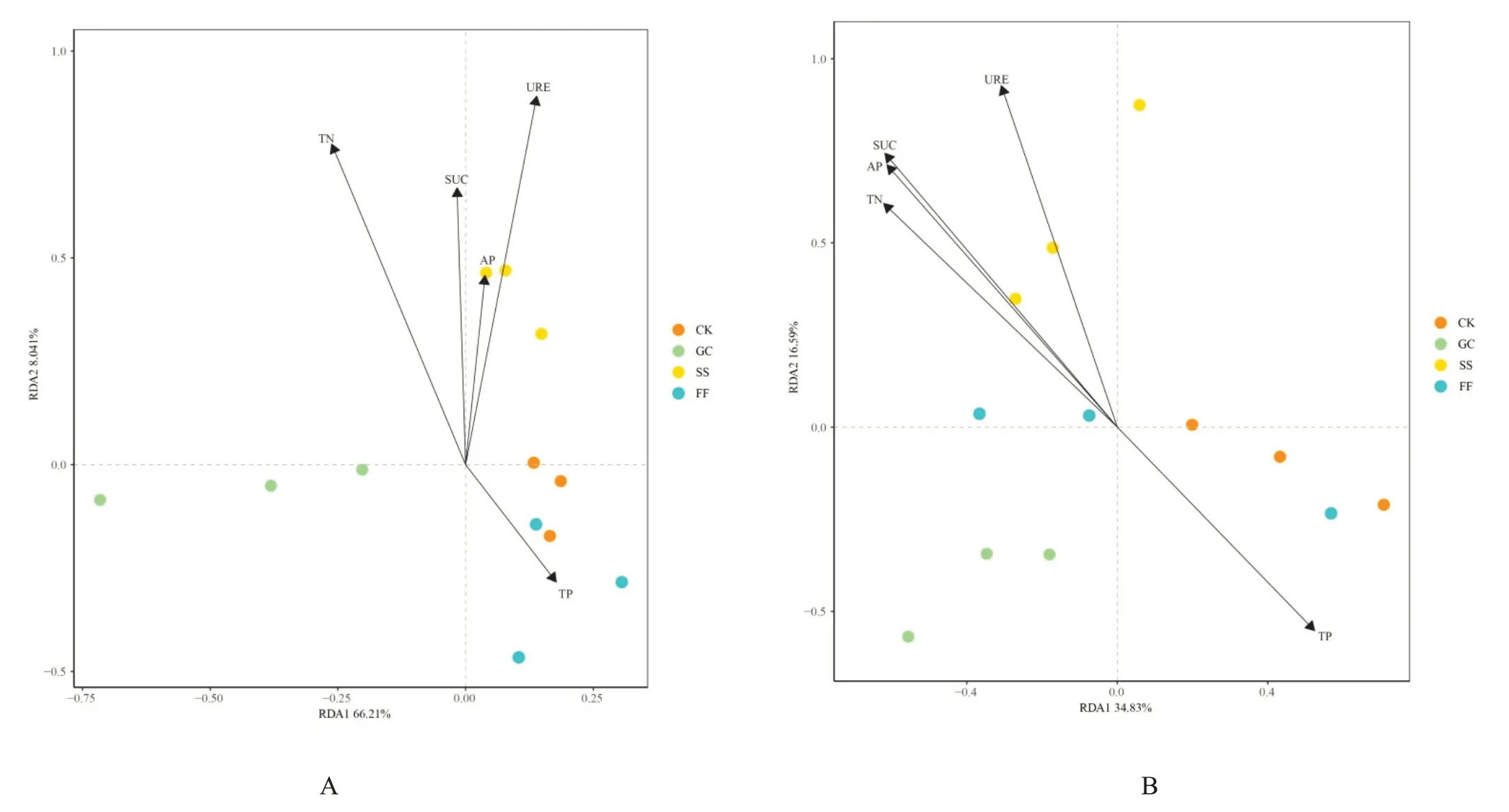
图3 优势细菌(A)和真菌(B)与土壤环境因子和植物类型的冗余分析
2.4 植物类型对土壤微生物群落的影响
方差分解分析植物在细菌门水平(图4A)和真菌科水平(图4B)的差异与土壤因子对植物根际微生物群落的相对大小,结果显示土壤和植物类型的交互作用对细菌群落差异的解释率为39.4%,而2个因素单独对细菌群落结构解释率均小于0。植物种类、土壤分别解释了3 种植物根际真菌群落差异的8.49%和1.61%,两因素交互共同解释率为9.75%。可见土壤环境因子与植物的交互作用对细菌群落结构有重要影响,而环境因子和植物类型均对真菌群落结构有明显的影响。
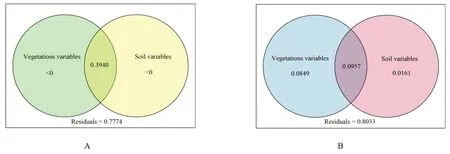
图4 方差分解分析土壤环境因子和植物类型对细菌(A)与真菌(B)群落影响相对大小
3 讨论与结论
在植物分类学上,甘草属于豆科(Fabaceae),防风和北沙参属于伞形科(Apiaceae)。本研究在微尺度局域空间上分析了3种药用植物根际和非根基土壤微生物群落结构。土壤理化性质分析结果表明,非根际土壤和根际土壤间全磷和过氧化氢酶的差异最为显著,其中在根际土壤中全磷含量较低而过氧化氢酶含量较高。已有研究表明过氧化氢酶与土壤中有机质含量呈正相关,可以作为土壤肥力的指标[25-26]。可见根际土壤的肥力更好,这可能与试验田肥料的施用有关。土壤群落分析结果表明,非根际土壤较根际土壤菌群丰富度较低。各样本菌群分布具有相似性,在细菌菌群群落中变形菌门和放线菌门的占比均较大,在真菌群落中赤壳科和微囊科的占比均较大。这与对内蒙道地药材黄芪的研究结果一致,在黄芪的研究中也发现放线菌门和变形菌门为优势菌门[27]。这可能因为变形菌门适应能力强,属于自然界中最普遍的菌门之一。变形菌门含有较多的固氮微生物,参与了重要的有机质转化过程和土壤结构形成过程[28]。豆科甘草区别于其他两种伞形科药用植物,其优势细菌群落为拟杆菌门,优势真菌群落为鳞毛蕨科和被孢霉科。已有研究表明,植物自身的掉落物优化了植物对氮和磷等稀缺养分的吸收,诱导了不同的外生菌根群落发育[29]。被孢霉科作为常见的优势真菌,对氮处理敏感[30]。土壤的有机质来源往往取决于地上植被凋落物的种类,而拟杆菌门在有机质分解和多糖代谢过程起着重要的作用[31]。由此推测,与伞形科植物不同,豆科的甘草含有的凋落物类型和氮素环境可能影响了甘草根际菌群的分布。
Spearman分析结果表明,环境因子对微生物群落的复杂程度影响明显,细菌菌门群落复杂程度与全氮、脲酶、蔗糖酶和酸性磷酸酶呈负相关,真菌菌科群落复杂程度与全钾和过氧化氢酶呈负相关。这与阿魏和玉米等植物根际菌群结构的研究结果一致,磷和钾等因子也驱动了其根际菌群结构[32-33]。但土壤有机质和氮磷钾等理化性质对土壤微生物群落的影响复杂,目前还没有完全一致的结论[27]。对优势细菌门和真菌科以及土壤理化性质的冗余分析,结果表明细菌和真菌群落的结构主要由全氮、全磷、脲酶、蔗糖酶和全磷驱动。驱动因子脲酶在塑造细菌和真菌群落功能特征方面起着重要作用,它与土壤有机碳和总氮呈高度相关,它与土壤有机碳和总氮呈高度相关,在氮循环中也发挥重要作用,同时提高土壤生态系统中的植物修复效率[34-35]。结合方差分解分析发现,土壤环境因子与植物的交互作用是细菌群落结构的主要驱动因素,而环境因子和植物类型对真菌群落结构均有影响,这与晋东南地区3种道地药材的研究结果一致[36]。该结果可能与真菌根际效应强、变异性高有关[37-38]。
综上所述,在试验田的局域环境中,根际土壤菌群中变形菌门和放线菌门是防风、甘草和北沙参的共有优势细菌菌门,占比分别在27.85%~52.69%和18.39%~44.59%之间;赤壳科和微囊科是共有的优势真菌菌科,占比分别在3.70%~14.94%和4.66%~30.32%之间。其中,甘草与其他两种伞形科植物在菌群结构上有较大差异,其优势细菌群落为拟杆菌门(18.86%),优势真菌群落为鳞毛蕨科(7.37%)和被孢霉科(9.41%),可能与其为豆科植物有关。土壤环境因子与植物种类单独对细菌群落结构的解释率均小于0,而两者交互作用对细菌群落差异的解释率为39.4%,可见土壤环境因子与植物种类的交互作用是细菌群落结构的主要驱动因素;植物种类和土壤环境因子分别解释了真菌群落差异的8.49%和1.61%,两者交互作用对真菌群落差异的解释率为9.75%,可见环境因子和植物类型均对真菌群落结构有影响。总体来说,植物—微生物—土壤间相互作用为药用植物构建了独特的生长环境。
猜你喜欢
中老年保健(2022年2期)2022-08-24
中国土壤与肥料(2021年5期)2021-12-02
科学(2020年4期)2020-11-26
中国比较医学杂志(2020年4期)2020-05-26
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
天然产物研究与开发(2018年3期)2018-05-07
中国蔬菜(2016年8期)2017-01-15
动物营养学报(2015年10期)2015-12-01
