
无碳氮化硼前驱体研究进展
2023-12-16 12:12:58沈绥卢振西段玉婷赵樱淼梁兵龙佳鹏
辽宁化工 2023年11期
沈绥 卢振西 段玉婷 赵樱淼 梁兵 龙佳鹏

摘 要:无碳氮化硼前驱体是前驱体法制备氮化硼陶瓷块体和基体材料的关键材料。介绍了聚硼吖嗪、聚氨基环硼氮烷、氨硼烷及其他类型的无碳氮化硼前驱体的制备方法、性能及研究现状,并对无碳氮化硼前驱体发展进行了展望。
关 键 词:氮化硼;无碳前驱体;聚硼吖嗪;聚氨基环硼氮烷;氨硼烷
中图分类号:TQ174.75+8.12 文献标识码: A 文章编号: 1004-0935(2023)11-1633-06
氮化硼陶瓷是一种人工合成陶瓷。1842年,BALMAIN[1]采用氧化硼与氰化钾反应,第一次制得氮化硼,但氮化硼陶瓷直到20世纪下半叶才得到广泛的应用。氮化硼主要有4种晶体结构:六方氮化硼(h-BN)、立方氮化硼(c-BN)、菱方氮化硼(r-BN)和纤锌矿氮化硼(w-BN),其中六方氮化硼是常规条件下的热稳定相。氮化硼陶瓷是密度最小的陶瓷,且具有耐高温、抗氧化、耐化学腐蚀、无毒色白、自润滑、加工性好、高温下与多种金属不浸润且具有良好的导热性、优异的介电性能以及透波特性等优良的性质[2],这些特点使得氮化硼材料在航空航天、中子防护、电子材料、环保等领域都具有广阔的应用前景。
氮化硼陶瓷的制备方法主要有高温热压法、化学气相沉积法和前驱体法。高温热压法是传统陶瓷制备方法,通常只能制备块状结构简单的制品。化学气相沉积生产效率低、成本高,通常只适于制备涂层、薄膜制品。前驱体法是20世纪60年代逐渐发展起来的新一代陶瓷制备方法,可制备不同结构、形状、形态的氮化硼材料(如纤维、薄膜或三维的特定形状)的产品。尤其是前驱体转化法制备的氮化硼基陶瓷复合材料,凭借密度最小、耐高温性能和介电性能优异,成为高超音速飞行器天线罩的首选材料[3-5]。前驱体转化法制备的氮化硼多孔材料比表面积大,硼氮化学键具有极性,内部活性点多,材料本身耐化学腐蚀、抗氧化、无毒,成为气体吸附和液体吸附新材料的研究热点[6-8]。
目前氮化硼前驱体主要为环硼氮烷聚合物,如B-氨基-N-烃基环硼氮烷聚合物、B-胺基环硼氮烷聚合物[9]、B-胺基-N-烷基环硼氮烷聚合物[10],这些前驱体聚合物在硼氮六元环上引入烷基或芳基,改善了前驱体的加工性能,可以制备纤维、涂层等。由于其比表面积大,在热解过程中采用氨气气氛可以有效脱除碳元素,得到含碳量很低的氮化硼。但是,这类含碳的先驱体制备氮化硼基体材料及块状材料时,材料内部的碳受扩散的影响,很难被高效脱除,这种情况要得到得到纯的BN材料,就需要氮化硼无碳前驱体。
目前科研工作者合成了大量不同结构的氮化硼前驱体,国内外很多文献也对氮化硼前驱体的研究进展进行了系统论述[11-13]。但对制备基体材料氮化硼或块状氮化硼材料的无碳前驱体研究进展,目前国内外都还没有专门的论述。本文在追溯硼氮化合物发展的基础上,重点概述了无碳氮化硼前驱体开发历史及最新研究进展。
目前,国内外主要研究的无碳前驱体有聚硼吖嗪、聚氨基环硼氮烷两类,有部分研究者也开展了氨硼烷作为氮化硼前驱体应用的尝试。除此之外,有些研究也在探索新型的氮化硼无碳前驱体。
1 硼吖嗪及聚硼吖嗪
1.1 硼吖嗪的结构和性质
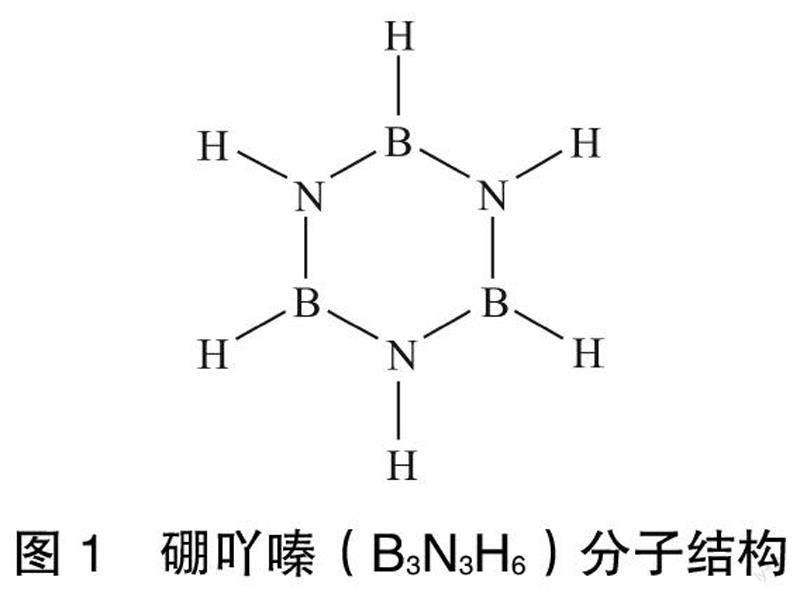
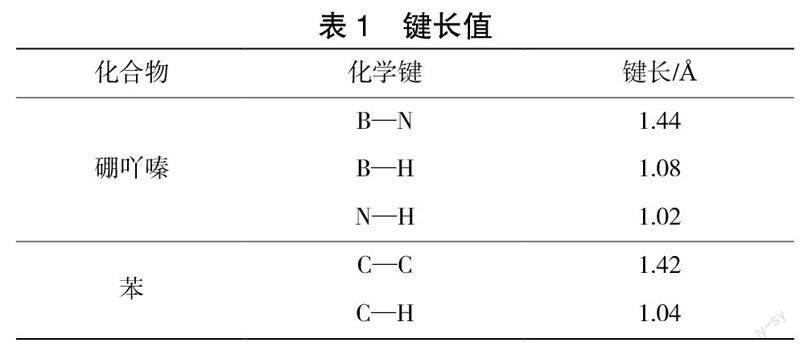
1926年,STOCK[14]等首次合成了硼氮六元环化合物硼吖嗪H3B3N3H3(全氢环硼氮烷)。硼吖嗪在常温下为无色液体,易挥发,熔点为-58 ℃,沸点为55 ℃,常温储存时尤其见光容易爆炸。硼吖嗪分子是苯的等电子体,其结构与苯相似,为平面结构,其分子结构如图1所示。硼吖嗪的键长与苯的键长也非常接近,见表1。
硼吖嗪只有硼氮氢3种元素组成,氢元素在热解过程中容易脱除;硼氮六元环的平面结构与h-BN的平面六元环结构一致,且B—N键的键长键角也相同,热解过程中,由硼吖嗪六元环向h-BN六元环演变容易,因此硼吖嗪的聚合物或硼吖嗪衍生物的聚合物作为氮化硼前驱体是优选结构。
1.2 硼吖嗪的合成工艺
1926年最早由STOCK[14]等公布合成硼吖嗪,其将乙硼烷与氨气在密闭容器中180~190 ℃加熱 3 h,通过热解[H2B(NH2)2+][BH4+]来合成硼吖嗪。
1954年SCHAEFFER[15]等则是通过将三氯环硼氮烷(TCB)溶液滴加到NaBH4溶液中,将TCB中的Cl原子还原为H原子进而得到硼吖嗪,反应式如式(1)所示。
3B3Cl3N3H3+3NaBH4+3(n-C4H9)3N
→B3N3H6+3NaCl+3(n-C4H9)3NBH3。 (1)
此方法步骤较多,合成路线与操作较为复杂,且会产生一氯环硼氮烷和二氯环硼氮烷等副产物。
1979年Callery Chemical 公司报道H3B·NH3溶液在100~160 ℃下热解生成硼吖嗪[16]。反应式如 式(2)所示。
3H3B·NH3→B3N3H6+6H2。 (2)
其反应原料氨硼烷常温下稳定,不与水反应,且易溶于水,原料贮存方便;但氨硼烷合成技术仍处于实验阶段,并未实现工业化生产,所以此方法目前并不能用来大量制备硼吖嗪。
1970年VOLKOV[17]等在前人的基础上,对硼氢化物和铵盐固相反应法进行了改良,将NaBH4与NH4Cl在230 ℃的高能量震动下进行固相反应得到硼吖嗪,反应式如式(3)所示。
3NaBH4+3NH4Cl→B3N3H6+3NaCl+9H2。 (3)
此方法虽然原料上更经济更安全,但产物中含有氯代硼吖嗪,且高温固相反应对实验条件要求 较高。
1995年SNEDDON[18]等将NaBH4与(NH4)2SO4在120~140 ℃溶液中进行反应,成功得到硼吖嗪,反应式如式(4)所示。
6NaBH4+3(NH4)2SO4→2B3N3H3+3Na2SO4+18H2。(4)
此方法无论是在原料成本还是操作难度与安全性上都更优于上述其他的方法,是目前制备硼吖嗪的优选方法。
但硼吖嗪熔点低,易挥发,加热不能得到氮化硼产物,不能直接作为分子前驱体。硼吖嗪是一种易挥发的液体,直接热解硼吖嗪得不到氮化硼陶瓷。要制备出氮化硼合适的前驱体,硼吖嗪要聚合为可熔或可溶、不挥发的聚合物。
1.3 聚硼吖嗪的聚合工藝及应用
1926年,STOCK[14]等在报道合成硼吖嗪的同时,也报道了硼吖嗪加热到500 ℃发生反应,得到氢气和不挥发的固体聚合物。
1961年,LAUBENGAYER[19]等报道了硼吖嗪热脱氢缩合反应的研究,硼吖嗪加热到340~440 ℃,反应生成了氢气、萘型稠环化合物B5N5H8、联苯型化合物B6N6H10、2,4-二胺基环硼氮烷等小分子化合物以及化学式为BNH0.8的不挥发聚合物。报道中还对比了硼吖嗪与苯热缩合反应的不同,苯热缩合过程中,几乎没有开环反应,而硼吖嗪热缩合反应发生了大量的开环反应。
1971年,NEISS[20]等报道了光催化气相硼吖嗪的热缩合反应,硼吖嗪在184.9 nm紫外线的照射下进行反应,得到了氢气、萘型的B5N5H8、微量的联苯型B6N6H10等小分子和不挥发的硼吖嗪聚合物固体。在小分子化合物中,B5N5H8较多而B6N6H10微量,证明了硼吖嗪光催化缩合易于开环反应。
20世纪80年代后期到90年代前期,SNEDDON等开展了硼吖嗪的聚合反应的研究,他们开始将反应条件设定为过渡金属催化剂作用下的热缩合,但在实验中发现,硼吖嗪在中等温度、真空条件下就可以自聚合[21-22]。SNEDDON等在70 ℃的温度、真空条件下,反应48 h得到硼吖嗪可溶的低聚物。延长时间或升高温度都会使得硼吖嗪过度交联,生成不溶不熔的白色产物,用此方法制备的氮化硼粉末其陶瓷产率可达到85%~93%。SNEDDON等还将无机纤维束浸在聚硼吖嗪的甘醇二甲醚溶液中,然后在氨或氩气气氛中加热纤维至900 ℃,得到了优异的氮化硼涂层。其中得到的氧化铝纤维上的涂层在SEM下显示出具有0.2~0.4 μm均匀致密的BN涂层。
1993年,KIM[23]等报道了在氮气气氛、加压条件下,制备出了流动性优异的硼吖嗪低聚物。它采用浸渍法,制备了致密的碳纤维增强的氮化硼基复合材料(C/BN)。C/BN复合材料相比碳材料大大提升了抗氧化能力,其抗氧化温度可达850 ℃,而碳材料的抗氧化温度只有400 ℃。
1998年,PARLIER[24]等公布了一种液氨引发硼吖嗪氨解聚合的新工艺,可大幅加速聚合过程,聚合时间从热聚合的48 h缩短到2 h,并将产物的溶液浸渍碳纤维,得到厚度0.1~1.2 μm且厚度可调的BN涂层。GERVAIS[25]等对氨引发的硼吖嗪聚合物与热引发的硼吖嗪聚合物及二者的热解过程进行了研究,其对比结果如表2所示。
由分析结果可知,氨引发的硼吖嗪聚合物只是氢元素含量高一些,而热解温度超过200 ℃后产物的元素含量几乎一致。
聚硼吖嗪中仍有大量活泼B—H、N—H键,尤其是B—H键非常活泼,造成聚硼吖嗪易燃易爆,使用过程条件控制严苛。而胺基环硼氮烷,硼氮六元环上B元素的H被—NH2取代,其安全性大幅提高,其聚合物聚胺基环硼氮烷是研究的另一个热点。
2 氨基环硼氮烷及聚氨基环硼氮烷
1959年,NIEDENZU[26]等用B-三氯环硼氮烷(TCB)与胺反应,制得一系列环硼氮烷取代产物。他们尝试用TCB与NH3(液氨或惰性溶剂中)反应,得到了白色沉淀,沉淀被推测是氨基环硼氮烷(及缩聚物)与NH4Cl的混合物,但他们分离出氨基环硼氮烷的尝试没有成功。
1961年,MIKHAILOV[27]等用B-三(正丁硫基)环硼氮烷氨解,得到了无色结晶沉淀三胺基环硼氮烷,其反应过程如式(5)所示。
得到的三胺基环硼氮烷不溶于苯、异戊烷和乙醚,加热到250 ℃不熔融,分解放出氨气。
1961年,NIEDENZU[28]等将无水NH3通入到 B-三(二甲胺基)环硼氮烷的惰性溶液中,得到了纯的胺基环硼氮烷白色沉淀。此产物结晶良好,对水汽非常敏感。
此后的近20年,由于当时缺乏明确的实际应用,环硼氮烷的研究进入了低潮期。到了20世纪80年代后期,由于前驱体转化法陶瓷制备的兴起(以碳化硅纤维产业化为代表),作为氮化硼陶瓷最有潜力前驱体的环硼氮烷,逐渐成为人们的研究热点。
20世纪80年代后半段到90年代初期,美国杜克大学的PAINE教授及其合作研究者[29],在桑迪亚国家实验室的资助下,采用TCB与六甲基二硅氮烷(HMD)反应的工艺路线,其反应式如式(6) 所示。
反应得到胺基环硼氮烷的凝胶。此产物不熔,可溶于液氨,在有机溶剂中不溶。他们对此聚合物进行热解,得到了较纯的BN。
TCB的环上的3个硼原子与3个氯原子相连,在与HMD反应时,会形成体形聚合物,难于熔融和溶解。PAINE[30]等为了提升前驱体的熔融及溶解性,尝试制备线性的聚胺基环硼氮烷,其工艺路线如式(7)所示。
他们对此产物热解后得到的BN,碳质量分 数<5%,但产物还是不能达到易溶或可熔。
1989年,NARULA[31]团队在300~400 ℃条件下对[(Et2N)BNH]3与[(Me2N)BNH]3进行氨解实验时发现,氨解产物中存在无碳氮化硼前驱体,但却难以将其从产物中分离出来。在此基础上,他们通过向[(Me2N)BNH]3的甲苯溶液中通入过量的NH3,对得到的白色固体产物进行了红外光谱和核磁共振分析,得出固体产物中可能存在低交联形态的胺基环硼氮烷。反应式如式(8)所示。
得到的无碳前驱体难以溶解和熔融,通过烧结可以较容易的得到氮化硼粉末,但是对于此前驱体的性能并没有详细报道。
1992年,KIMURA[32]团队将B-三(二乙胺基)環硼氮烷(TDEAB)在压力7~8 atm、温度-78 ℃的条件下与液氨进行反应,反应式如式(9)所示。
再经高温减压升华提纯后成功制备出三甲胺基环硼氮烷(TAB)单体结晶(白色晶体粉末,不溶解,微溶于芳香族溶剂)。他们对TAB进行了热聚合实验,通过计算热解过程中TAB失重率发现,控制反应温度和时间可以得到不完整的BN网络结构,也就是TAB的低聚物前驱体。TAB与其低聚物前驱体均可在惰性气氛中通过热解直接转化为BN粉末,通过对团状TAB和TAB低聚物前驱体的烧结对比实验发现,TAB低聚物的烧结产物性能明显优于TAB单体,但是TAB低聚物前驱体的加工性能很差。
3 氨硼烷
氨硼烷(NH3BH3)是由硼原子与氮原子配位键相结合的配位化合物,常温常压下为白色固体,无毒,在空气中稳定,易溶于水,加热放出氢气,目前被视为最具潜力的储氢材料之一[33]。氨硼烷只含有硼氮氢元素,且其加热可以部分转化为硼吖嗪,继而形成聚硼吖嗪,高温处理后可得到BN陶瓷,因此,也是一种潜在的氮化硼前驱体。
氨硼烷最早是于1955年由 SHORE[34]等合成,他们用两条合成路线得到了氨硼烷,一种是将LiBH4与氯化铵或硫酸铵在溶剂中反应生成氨硼烷,如式(10)、式(11)所示;另一种是将乙硼烷二铵与氯化铵在微量NH3的环境下反应得到氨硼烷,如式(12)所示。
LiBH4+NH4Cl→LiCl+H3NBH3+H2。 (10)
2LiBH4+(NH4)2SO4→Li2SO4+2H3NBH3+2H2。(11)
[H2B(NH3)2+][BH4-]+NH4Cl
→[H2B(NH3)2+][Cl-]+H3NBH3+H2。 (12)
目前,研究者们合成氨硼烷的主要路线有两条:一条是由氨分子取代不同硼烷络合物中的弱路易斯碱(L)生成氨硼烷,反应式如式(13)所示;另一条是碱金属硼氢物与铵盐的置换反应获得,反应式如式(14)所示。
L·BH3+NH3→NH3BH3+L。 (13)
BH4-+NH4+→NH3BH3+H2。 (14)
2011年,钟博等[35]将氨硼烷作为制备氮化硼陶瓷的前驱体并成功制备出BN纳米管、BN纤维等氮化硼材料。因此用氨硼烷作为无碳前驱体制备氮化硼陶瓷极具潜力且具有较为宽广的应用前景。
4 其他无碳氮化硼前驱体
1999年,KIM[36]等在对氨硼烷的连续脱氢热解实验中发现,脱氢热解反应中会产生一种链状结构的硼氮烷中间体(NH2BH2)x,这种中间体之后会迅速反应生成B3N3H6和聚胺基硼烷,反应式如式(15)所示。
实际上,早在1995年WIDEMAN[37]等曾报道过NH2BH2单体会在-196 ~ -155 ℃之间自发聚合,转化为聚氨基硼烷(NH2BH2)x。因此以(NH2BH2)x作为无碳氮化硼前驱体,具有一定的研究价值。
2021年,杜贻昂[38]等发明了一种由多元硼氮桥连接六元环结构的无碳先驱体,该前驱体具有高陶瓷产率、相对稳定性好、无杂质等特点。该方法首先将二(三甲基硅基)氨基钠(NaHMDS)与氨硼烷(AB)在30~90 ℃下搅拌12~48 h,之后将过滤后的固体与NH3Cl以二甲氧基乙烷为溶剂在30~90 ℃下搅拌12~48 h,过滤干燥后得到白色的低聚氨硼烷。最后将低聚氨硼烷与三氯环硼氮烷(TCB)加入到溶剂中,在30~90 ℃下搅拌12~48 h后真空蒸干溶剂后得到该无碳氮化硼前驱体。反应式如 式(16)至式(18)所示。
杜贻昂等对此方法得到的无碳氮化硼前驱体进行烧结,结论显示通过调节反应物的量与反应条件,得到的无碳氮化硼前驱体最优陶瓷产率可达82%。
5 结论和展望
聚硼吖嗪陶瓷產率高,可熔可溶,是目前已开发最合适的氮化硼无碳前驱体。但聚硼吖嗪需在低温下保存,存放运输都不方便,且易于爆炸,使用条件苛刻,安全系数低。聚胺基环硼氮烷相较于聚硼吖嗪,化学性质稳定,但目前合成的产品不熔,在有机溶剂中不溶或溶解性不好,限制了其作为前驱体的应用。氨硼烷和其他类型的无碳前驱体作为氮化硼的前驱体尚在初期探索阶段,未有成功应用的报道。
下一步,开发安全的无机基团改性聚硼吖嗪,或开发安全的聚硼吖嗪储运技术是研究方向之一;通过结构设计,提升其他无碳前躯体可熔或可溶性能,是研究的另一方向。
参考文献:
[1]BALMAIN W H. Bemerkungen über die Bildung von Verbindungen des Bors und Siliciums mit Stickstoff und gewissen Metallen[J]. Journal Für Praktische Chemie, 1842, 27(1):422-430.
[2]邓橙. 氮化硼纤维先驱体—聚硼氮烷的合成及热解特性研究[D]. 长沙:国防科学技术大学,2009.
[3]周韵宁.陶瓷基复合材料天线罩制备工艺的研究[J].陶瓷,2021(6):97-98.
[4]高龙飞,柴笑笑,马君毅,等.石英纤维增强氮化硼陶瓷基复合材料制备及性能研究[J].复合材料科学与工程,2020(10):101-104.
[5]KANDI K K, PUNUGUPATI G, PAGIDI M, et al. A novel gelcast SiO2-Si3N4-BN ceramic composites for radome applications[J]. Silicon, 2022:1-14.
[6]LEI W, PORTEHAULT D, LIU D, et al. Porous boron nitride nanosheets for effective water cleaning[J]. Nat Commun, 2013, 4: 1777.
[7]赵召. 多孔六方氮化硼基纳米材料可控制备及吸附性能研究[D].西安:西安科技大学,2021.
[8]何适,李杰,戴伟,等.多孔氮化硼复合水净化材料的制备及其再生性能[J].武汉工程大学学报,2019,41(6):553-558.
[9]李宗鹏,王长松,梁兵.以三(二乙胺基)环硼氮烷为前驱体制备六方氮化硼[J].沈阳化工大学学报,2020,34(1):25-30.
[10]CORNU D, BERNARD S, DUPERRIER S, et al. Alkylamino- borazine-based precursors for the preparation of boron nitride fibers by the polymer-derived ceramics (PDCs) route[J]. Journal of the European Ceramic Society, 2005, 25(2-3):111-121.
[11]周瑩莹,张昭环.有机前驱体法制备氮化硼纤维的研究进展[J].合成纤维,2013,42(6):35-38.
[12]薛晓琳,王长松,梁兵.前驱体法热解合成氮化硼的研究进展[J].化工新型材料,2016,44(1):1-3..
[13]BERNARD S, SALAMEH C, MIELE P. Boron nitride ceramics from molecular precursors: synthesis, properties and applications[J]. Dalton Trans., 2016,45:861-873.
[14]STOCK A, POHLAND E. Borwasserstoffe, IX.: B3N3H6[J]. Berichte der deutschen chemischen Gesellschaft (A and B Series), 1926, 59(9):2215 - 2223.
[15]SCHAEFFER R, STEINDLER M, HOHNSTEDT L, et al. Prepara- tion of Borazole by the Reduction of Trichloroborazole1a[J]. Journal of the American Chemical Society, 1954, 76(12):3303-3306.
[16]HOUGH W V, GUIBERT C R, HEFFERAN G T. Method for the synthesis of borazine: US, US4150097A[P]. 1979-04-17.
[17]VOLKOV V. Preparation of borazine by the reaction of sodium tetrah- ydroborate wht ammonium chloride[J]. J. Inorg. Chem, 1970, 15: 1510-1513.
[18]WIDEMAN T, SNEDDON L G. Convenient procedures for the laboratory preparation of borazine[J]. Inorganic Chemistry, 1995, 34(4):1002- 1003.
[19]LAUBENGAYER A W, MOEWS P C, PORTER R F. The condensation of borazine to polycyclic boron-nitrogen frameworks by pyrolytic dehydrogenation1[J]. Journal of the American Chemical Society, 1961, 83(6):1337-1342.
[20]NEISS M A, PORTER R F. Photochemistry of borazine. Evidence for a borazyne intermediate[J]. Journal of the American Chemical Society, 1972, 94(5):1438-1443.
[21]PAINE R T, SNEDDON L G. Recent developments in borazine-based polymers[J].ACS Symp Series, 1994, 27:358-374.
[22]SNEDDON L G, MIRABELLI M, LYNCH A T, et al. Polymeric precursors to boron based ceramics[J]. Pure & Applied Chemistry, 1991, 63(3):407-410.
[23]KIM D P, ECONOMY J. Fabrication of oxidation-resistant carbon fiber/ boron nitride matrix composites[J]. Chemistry of materials, 1993, 5(9): 1216.
[24]PARLIER M, ROPARS M, VAULTIER M, et al. Textile preforms sheathed in a boron nitride coating, composite incorporating them and their preparation[J]. World Patent, 1998, 98:29355.
[25]GERVAIS C, FRAMERY E, DURIEZ C,et al. 11B and 15N solid state NMR investigation of a boron nitride preceramic polymer prepared by ammonolysis of borazine[J]. Journal of the European Ceramic Society,2005,25:129-135
[26]NIEDENZU K, DAWSON J W. Boron-nitrogen compounds. I. syntheses of B-aminoborazines1[J]. Journal of the American Chemical Society, 1959, 81(14):3561-3564.
[27]MIKHAILOV B M, Galkin A F. Synthesis and properties of B-tri- n-butylmercaptoborazoles[J]. Russian Chemical Bulletin, 1961(10): 345-347.
[28]NIEDENZU K, DAWSON J W. B-triaminound B-trihydrazino- borazol[J]. Angewandte Chemie, 1961, 73(12): 433.
[29]NARULA C K, SCHAEFFER R, PAINE R T, et al. Synthesis of boron nitride ceramics from poly(borazinylamine) precursors[J]. Journal of the American Chemical Society, 1987, 109(18): 5556-5557.
[30]PAINE R T, NARULA C K. Synthetic routes to boron nitride[J]. Chemical Reviews, 1990, 90(1):73-91.
[31]NARULA C K, SCHAEFFER R, DATYE A, et al. Synthesis of boron nitride ceramics from 2,4,6-triaminoborazine[J]. Cheminform, 1990, 21(21): 4053-4055.
[32]KIMURA Y, KUBO Y, HAYASHI N. Boron nitride preceramics based on B,B,B-triaminoborazine[J]. Journal of Inorganic and Organo- metallic Polymers, 1992, 2(2):231-242.
[33]鄧霁峰,陈顺鹏,武晓娟,等.水解制氢材料研究进展[J].无机材料学报,2021,36(1):1-8.
[34]SHORE S G, PARRY R W. The crystalline compound ammonia-borane, NH3BH3[J].Journal of the American Chemical Society, 1955, 77(22): 6084-6085.
[35]鐘博. 以氨硼烷为先驱体制备BN微纳米材料及其机理研究[D].哈尔滨:哈尔滨工业大学,2011.
[36]KIM D P, MOON K T, KHO J G, et al. Synthesis and characterization of poly(aminoborane) as a new boron nitride precursor[J]. Polymers for Advanced Technologies, 1999, 10:702-712.
[37]WIDEMAN T, SNEDDON L G. Convenient procedures for the laboratory preparation of borazine[J]. Inorganic Chemistry, 1995, 34(4):1002-1003.
[38]杜贻昂,王兵,王应德.一种高陶瓷产率的无碳氮化硼先驱体及其合成方法:CN113716581A[P].2021-11-30.
Research Progress of Carbon-free Boron Nitride Precursors
SHEN Sui, LU Zhen-Xi, DUAN Yu-ting, ZHAO Ying-miao, LIANG Bing, LONG Jia-peng
(Shenyang University of Chemical Technology, Shenyang Liaoning 110000, China)
Abstract: The boron nitride precursor without carbon is the key material for the preparation of boron nitride ceramic blocks and matrix materials by the precursor method. The preparation methods, properties and research status of poly boracazine, poly aminocyclic borazane, amborane and other types of carbon-free boron nitride precursors were introduced. The development trend of carbon-free boron nitride precursors was also prospected.
Key words: Boron nitride; Carbon-free precursor; Polyborazine; Polyaminocyclic borazane; Ammonia borane
猜你喜欢
化工设计通讯(2024年1期)2024-04-08 02:50:52
制造技术与机床(2017年10期)2017-11-28 05:20:15
材料科学与工程学报(2016年1期)2017-01-15 13:33:52
当代化工研究(2016年7期)2016-03-20 16:21:54
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:57
原子与分子物理学报(2015年3期)2015-11-24 12:49:39
化工进展(2015年3期)2015-11-11 09:08:56
电源技术(2015年9期)2015-06-05 09:36:06
物理化学学报(2015年5期)2015-02-28 17:34:53
应用化工(2014年11期)2014-08-16 15:59:13
