
羊栖菜水提物-鱼明胶复凝聚反应表征及其应用
2023-12-15 04:32谭棋张名位马永轩郝娟张瑞芬池建伟黄菲贾栩超董丽红马勤邓梅赵东
现代食品科技 2023年11期
谭棋,张名位,马永轩,郝娟,张瑞芬,池建伟,黄菲,贾栩超,董丽红,马勤,邓梅,赵东
(1.福建农林大学食品科学学院,福建福州 350002)(2.广东省农业科学院蚕业与农产品加工研究所,农业农村部功能食品重点实验室,广东省农产品加工重点实验室,广东广州 510610)
羊栖菜(Sargassumfusiforme)是马尾藻科、马尾藻属的一种海藻植物[1],广泛分布于我国沿海,北起辽东半岛南至雷州半岛,其中浙江沿海养植规模最大[2]。羊栖菜富含蛋白质、多糖、多酚等营养物质[3],根据《神农本草经》和《本草纲目》记载,其具有“瘿瘤结气、水气浮肿、宿食不消”等功效,并且现代药理学研究也证实羊栖菜具有降血脂、抗血栓、缓解疲劳、增强免疫力等药理作用,是一种很好的药食两用植物[4]。它作为我国重要经济藻类,国人食用较少,主要出口到日本,深受消费者欢迎,被誉为“长寿菜”。目前,由于羊栖菜的加工方式较为简单,食用和入药方式主要是凉拌、高温煮熟等方式食用,因此研发新的羊栖菜加工和利用方法是非常必要的。
近年来,羊栖菜的活性物质受到了广泛研究,并证实其水提物中的多糖是其生物活性的主要成分,含量丰富,占总提取物的70%以上[5]。羊栖菜多糖具有调节免疫[6]、抗氧化[7]、降血脂[8]等生物活性,是一种很好的天然功能活性物质。同时,羊栖菜多糖作为天然的生物大分子,其分子中含有大量的羧酸根、硫酸根和醇羟基,表面带有大量的负电荷[9],可与携带相反电荷的大分子相互交联形成复聚物微胶囊壳,能够有效提升不稳定营养素的稳定性和生物利用率,例如花色苷,其化学性质不稳定,容易受温度、pH值、金属离子等因素影响,导致结构改变而失活[10]。
因此,本文为了探究羊栖菜水提物(SFE)的封装特性,利用常温超高压法制备的SFE,并选用应用广泛且具有生物兼容性的鱼明胶(FG)与之相互作用,通过探究浊度、Zeta电位和粒径等变化规律,明确pH值和壁材比对SFE与FG复合凝聚反应的影响因素,并验证二者复合凝聚制备花色苷微胶囊的应用潜力。
1 材料与方法
1.1 材料与试剂
羊栖菜购于浙江洞头岛,经-20 ℃冷链运输,储存于-18 ℃冷库;鱼明胶购于Sigma公司;大豆油购于阿拉丁试剂有限公司;赤峰籼稻黑米购于山东鹤来香食品有限公司;聚甘油蓖麻醇酸酯(Polyglycerol Polyricinoleate,PGPR)购于源叶生物科技有限公司;HCl购于广州化学试剂厂;NaOH购于大茂化学试剂厂;水为去离子水。
1.2 仪器与设备
TU-1900紫外可见分光光度计,北京普析通用仪器有限责任公司;Ultrasonic Cleaner SB25-12超声清洗机,宁波新芝生物科技有限公司;Ultra Turrax T-25Basic高速剪切机,德国IKA仪器有限公司;FGn5酶标仪,美国BioTek公司;Zetasizer Nano电位仪,英国Malvern公司;FDU-2110真空冷冻干燥机,东京理化器械株式会社;VERTEX 70傅里叶变换红外光谱仪,德国Bruker公司;恒温磁力搅拌器,上海湃澜仪器设备有限公司。
1.3 试验方法
1.3.1 花色苷的制备
根据Zhang等[11]的方法并稍作修改,具体步骤为:将黑米皮浸入酸化甲醇(甲醇:1 mol/L HCl=85:15,V/V)按料液比1:30(m/V),10000 r/min冰浴均质5 min,5000 r/min离心10 min,收集上清液,重复提取2次。合并3次离心上清,在45 ℃真空条件下旋蒸浓缩,再加水复溶,经-80 ℃预冻后,经真空冻干制成花色苷粉末,储存于-20 ℃冰箱备用。
1.3.2 羊栖菜水提物(SFE)的制备
羊栖菜浸泡于水中脱盐3 h、洗净除杂备用。将500 g羊栖菜(1:8.6,m/V)加去离子水,置于匀浆机中匀浆5 min,匀浆结束后分装于铝箔袋中密封,选择保压压力400 MPa和保压时间15 min的条件对羊栖菜浆液进行超高压处理,再将其4000 r/min离心20 min收集上清液,冻干后得到SFE。
1.3.3 羊栖菜水提物-明胶(SFE-FG)复聚物的制备
根据Fataneh等[12]的方法并稍作修改,准确称取一定质量的SFE、FG样品溶于去离子水中,在室温条件下300 r/min磁力搅拌2 h后放置4 ℃冰箱水合过夜,得到1%(m/V)SFE母液、2%(m/V)FG母液。将上述母液以SFE与FG质量比分别为1:3、1:2、1:1、2:1和3:1且复聚物总质量分数为0.1%(m/V)混合。室温下,SFE-FG混合液在300 r/min磁力搅拌6 h,以确保SFE和FG分子之间充分的静电相互作用。
1.3.4 SFE-FG复聚物的表征
1.3.4.1 Zeta电位测定
参考María等[13]的方法并稍作修改,分别将500 mL的0.1%(m/V)SFE溶液、0.1%(m/V)FG溶液、0.1%(m/V)不同质量比的SFE-FG混合液置于300 r/min磁力搅拌器中,分别用1 mol/L HCl或1 mol/L NaOH调节溶液pH值2至pH值10,在25 ℃恒温条件下,用Malvern Nano ZS电位仪测定体系的Zeta电位变化情况。
1.3.4.2 浊度测定
参考Lin等[14]方法并稍作修改,分别将500 mL的0.1%(m/V)不同质量比的SFE-FG混合溶液置于300 r/min磁力搅拌器中,分别用1 mol/L HCl和1 mol/L NaOH调节溶液pH值(1.0~7.0),采用酶标仪测定在600 nm下溶液的吸光度,绘制各溶液的浊度随pH变化的浊度曲线。用下式计算浊度:
式中:
T——浊度,cm-1;
It——透射光强度;
I0——入射光强度。
1.3.4.3 粒径测定
参考Zhang等[15]的方法并稍作改进,将500 mL 0.1%(m/V)SFE-FG混合溶液(SFE与FG质量比为1:3)置于300 r/min磁力搅拌器中,分别用1 mol/L HCl和1 mol/L NaOH调节溶液pH值(1~7),以蒸馏水(折射系数为1.33)为稀释溶剂,数据采集时间设定为180 s,动态采集时间为10 s,温度维持在25 ℃,用Malvern Nano ZS仪测定复聚物的粒径分布和多分散性指数(PDI)。
1.3.4.4 产率测定
参考Silva等[16]的方法并稍作修改,取50 mL 0.1%(m/V)不同质量比的SFE-FG混合溶液置于100 mL离心管中,在100 r/min磁力搅拌器条件下,分别用1 mol/L HCl和1 mol/L NaOH调节pH至最大浊度,并于5000 r/min离心15 min,收集沉淀于50 ℃烘干至恒重。通过下式计算复聚物产率:
式中:
F——复聚物产率,%;
m——干燥后复聚物质量,g;
m0——初始加入材料总质量,g。
1.3.4.5 微观结构分析
分别将50 mL 0.1%(m/V)不同质量比的SFE-FG混合溶液置于100 mL离心管中,在100 r/min磁力搅拌器条件下,分别用1 mol/L HCl和1 mol/L NaOH调节pH值至各比例的最大浊度对应的pH值(SFE与FG质量比分别为1:3、1:2、1:1、2:1和3:1时对应的pH值为4.2、3.4、2.6、1.6和1.2),取10 µL SFE-FG复聚物溶液于载玻片上,用倒置显微镜以400倍放大倍数观察。
1.3.4.6 傅里叶红外光谱分析
参考Anvari等[17]的方法稍作修改,分别称取1 mg SFE、FG和SFE-FG冻干粉末样品,加入100 mg的KBr进行研磨,再经压片机压制成片,选用纯KBr片作为空白对照。置于红外光谱仪上,设定在400~4000 cm-1范围内进行扫描,扫描次数为32次,分辨率为4 cm-1。
1.3.5 SFE-FG复合凝聚反应制备黑米皮花色苷微胶囊
参考Frank等[18]和Shaddel等[19]的方法并稍作修改,制备花色苷微胶囊。在室温下,将10%(m/V)花色苷溶液逐滴加入到含有3%(V/V)PGPR的大豆油预混合,用Ultra-Turrax T25高速分散器以12500 r/min均质5 min,制备油包水(W1/O)初级乳液(芯材)。再将初级乳液加入不同浓度的FG溶液,10000 r/min均质机中均质4 min,制备W1/O/W2双重乳液。向双重乳液中加入相应浓度的SFE溶液于100 r/min磁力搅拌器中搅拌6 h,用1 mol/L HCl调溶液pH值至4.2,再经真空冻干,即制得微胶囊。
1.3.6 黑米皮花色苷微胶囊包埋率的测定
1.3.6.1 花色苷含量的测定
采用pH示差法测定总花色苷含量,参考Wolfe等[20]的方法并稍作修改。分别取1 mL花色苷提取液于2个离心管中,分别加入1 mL KCl缓冲液(0.025 mol/L,pH值1.0)和CH3COONa缓冲液(0.4 mol/L,pH值4.5)到2个离心管中,混匀后避光、静置反应15 min,用紫外分光光度计分别在520 nm和700 nm波长测定样品的吸光度值,根据公式算出总花色苷含量:
式中:
C——总花色苷含量,mg/100 g;
A——测定的吸光度值;
e——矢车菊素-3-葡萄糖苷的摩尔吸光系数,26900 L/(cm·mol);
L——光径,1 cm;
M——矢车菊素-3-葡萄糖苷的分子量,449.2 g/mol;
D——稀释倍数;
V——最终体积,mL;
W——样品质量,g。
1.3.6.2 包埋率测定
参考Shaddel等[21]和Ji[22]的方法并稍做修改,测定花色苷包埋率。总花色苷含量:取100 mg微胶囊加入2 mL超纯水,研磨破坏微胶囊膜,再加入6 mL乙醇,于超声破碎,4000 r/min离心5 min,测定上清液花色苷含量。表面花色苷含量:取100 mg微胶囊加入2 mL超纯水和6 mL乙醇,100 r/min磁力搅拌5 min,3000 r/min离心4 min,测定上清液花色苷含量,通过下式计算包埋率:
式中:
Q——黑米花色苷包埋率,%;
M1——黑米花色苷总质量,mg;
M2——微胶囊外部的黑米花色苷质量,mg。
1.3.7 统计分析
除红外光谱实验以外,所有实验均重复3次,结果以平均值±标准差表示。数据处理及分析用SPSS 22软件,显著性分析采用方差分析(ANOVA)中的Duncan’s分析,显著性水平为0.05,用不同的字母表示;绘图用Origin 2017软件。
2 结果与讨论
2.1 SFE-FG复聚物的Zeta电位
生物大分子复合凝聚反应的主要驱动力源自其表面分布的不同电荷相互吸引,且其表面带电量与溶液pH值密切相关[23,24]。从图1可知,SFE的Zeta电位随pH值的升高而降低,且保持其Zeta电位为负值,最大为-58.86 mV。FG的Zeta电位也随着pH值的升高从16.65 mV下降到-5.41 mV,其等电点约为pH值8.5。在分散体系中,溶液的Zeta电位的绝对值越大,表明粒子之间的静电斥力作用越强,越不易出现凝聚现象[25]。因此,初步判定:pH值在2.0~8.5范围内,SFE与FG具有相反电荷,两种分子之间具有形成复聚物的潜力。

图1 SFE、FG溶液和SFE-FG复聚物(总质量分数为0.1%,m/V)的Zeta电位变化Fig.1 The change of zeta potential in SFE, FG and SFE-FG coacervate solutions (total mass concentration 0.1%, m/V)
本实验明确了SFE和FG两种大分子的Zeta电位与pH值之间的关系,并进一步探究了SFE和FG不同质量比混合溶液电位与pH的关系,进而明确了SFE-FG复聚物形成最佳比例及合适反应的pH值。由图1可知,pH值变化范围为2.0~10.0时,SFE-FG复聚物质量比为1:1、2:1和3:1的复合溶液的Zeta电位值均保持负值,且随pH值的升高,Zeta电位的绝对值变大,这是由于体系带负电荷的SFE主导了溶液的Zeta电位值,表明溶液中负电荷的过剩未能完全反应[26]。然而SFE-FG复聚物质量比为1:2、1:3,pH值在3和3.5附近时,体系中Zeta电位为0 mV,此时SFE分子所携带的负电恰好与FG分子所携带的正电完全中和。
2.2 SFE-FG复聚物的浊度曲线
通常蛋白质-多糖复聚物的复合凝聚反应过程可用浊度滴定法进行追踪[27],通过调整反应过程中的pH值可辨析反应进度。图2表示不同质量比的SFE-FG复聚物在不同pH值诱导下形成的浊度曲线。通过pH值可将浊度曲线分为共溶区、可溶性复聚物区、不可溶性复聚物区和共溶区四部分[28]。由图2中SFE-FG(1:3)可知,在这四个区域的分界处存在四个临界pH值(pHc值5.6、pHφ1值5.2、pHopt值4.2、pHφ2值1.8)。当pH 7≥pH≥pHc时,为SFE与FG的共溶区,此时浊度曲线趋平。当pHc>pH≥pHφ1时,为可溶性复聚物区,此时溶液浊度值随pH值降低而缓慢上升。当pHφ1>pH≥pHφ2时,为不可溶性复聚物区,此时溶液逐渐出现不溶性复聚物,浊度值随pH值降低出现迅速上升后下降现象,其中pH=pHopt时,浊度出现最大值,表明此时生成了最大的不溶性复聚物。当pH<pHφ2时,为共溶区,此时不溶性复聚物解离完全,浊度曲线趋平。Aryee和Nickerson[29]研究了扁豆蛋白分离物-阿拉伯树胶多糖复聚物的形成,并报道了相似的浊度曲线。因此,从浊度曲线中的临界pH点可以初步确定SFE和FG之间的复合凝聚反应的过程[24]。

图2 不同质量比SFE-FG复聚物(总质量分数为0.1%,m/V)的浊度曲线Fig.2 Turbidity of SFE-FG coacervate (total mass concentration 0.1%, m/V) prepared at different mass ratio
由图2可知,当SFE-FG复聚物质量比由1:1向1:2、1:3变化时,浊度曲线向右移动(向高pH方向),pHopt从2.6到4.2,且浊度值从0.74到0.95,这是因为FG的增加将导致SFE能结合的氨基数量增加,有利于形成不可溶性复聚物[30]。当SFE-FG复聚物质量比由1:1向2:1、3:1变化时,浊度曲线向左移动(向低pH方向),浊度值从0.74到0.34。乳清分离蛋白-木瓜种子黏液[31]、田间豌豆分离蛋白-壳聚糖[32]等体系也表现出相似的浊度曲线。此外,SFE-FG复聚物的浊度规律与前文Zeta电位所得结果一致。因此,在pH值1.0~7.0范围内,SFE-FG复聚物最佳比例为1:3,当pH值为4.2时,最大浊度值为0.95,表明最大的SFE-FG不溶性复聚物生成。
2.3 SFE-FG复聚物的粒径分布
反应体系中复聚物的粒径变化也是多糖与蛋白质静电相互作用的重要指标之一[33]。如表1所示,SFE和FG的平均粒径分别为6037.22 nm和607.10 nm。在pH值1.0~7.0范围内,SFE-FG复聚物的平均粒径出现先升高后降低的趋势。当pH在pHopt附近(pH值4)时,SFE-FG复聚物的平均粒径最大,为5080.67 nm。这是因为在不同pH值条件下多糖与蛋白质逐渐聚合形成大小不一的复聚物。随着pH值进一步降低(pH值<4),SFE-FG复聚物粒径逐渐变小,可能是不可溶性复聚物发生解离所引起的[34]。霍云峰[35]在研究卵清蛋白与坛紫菜多糖在不同pH值下的粒径分布时,发现平均粒径随pH值降低出现先升高后降低,最大平均粒径也在pHopt附近,这与本文SFE-FG复聚物的粒径分布结论类似。

表1 SFE-FG(1∶3)复聚物的平均粒径Table 1 Average particle size of SFE-FG coacervate (1:3)
2.4 SFE-FG复聚物的产率
复聚物产率反映了复合凝聚过程中微胶囊壁材的利用率[36]。不同质量比的SFE-FG复聚物会影响两种物质在溶液中的相对电荷密度,从而影响两种物质的静电相互作用,复聚物产率也会受到相应影响。从图3中可知,SFE-FG复聚物质量比例由1:1到1:3时,复聚物产率提高,这与Ardestani等[37]研究酪蛋白酸钠-果胶复聚物产率的结果类似,推测可能是由于静电相互作用的加强,导致可溶性复聚物减少,不溶性复聚物增多。当SFE-FG复聚物质量比为1:3时,复聚物产率最高达83.73%。可能是FG携带的正电荷氨基基团与SFE携带的负电荷硫酸根、羧基基团充分结合,中和体系中的电荷,发生最大的静电相互作用[26]。当SFE-FG复聚物质量比由1:1到3:1时,复聚物产率逐渐降低。可能是体系中SFE携带的负电荷远多于带FG携带的正电荷,产生更多的静电斥力,使得复聚物解离为水溶性而无法析出[38]。因此,选择SFE-FG复聚物质量比为1:3,pH值4.2为最佳的复合凝聚微胶囊壁材制备条件进行下一步研究。

图3 不同质量比SFE-FG复聚物(总质量分数0.1%,m/V)的产率Fig.3 Productivity of SFE-FG coacervate (total concentration 0.1%, m/V) prepared at different mass ratio
2.5 SFE-FG复聚物的显微结构
通过光学显微镜观察到SFE-FG复聚物质量比分别为1:3、1:2、1:1、2:1和3:1时对应的pH值为4.2、3.4、2.6、1.6和1.2(总质量分数为0.1%,m/V)所形成的复聚物的结构。由图4可知,SFE-FG复聚物质量比为2:1、3:1时,复聚物结构疏松且颗粒较小,该结果与前文浊度变化一致,此时溶液浊度较低。当SFE-FG复聚物质量比由1:1向1:2、1:3变化时,复聚物结构更为紧密,聚合成团现象明显,呈块状复聚物[34]。

图4 不同质量比SFE-FG(总质量分数0.1%,m/V)复聚物的形态Fig.4 Morphology of SFE-FG coacervate (total concentration 0.1%, m/V) prepared at different mass ratio
2.6 SFE-FG复聚物的红外光谱分析
图5 中为FG(曲线a)、SFE(曲线b)和不同比例SFE-FG复聚物(曲线c~g)的红外光谱分析图。FG在3446 cm-1呈现较宽的峰形,主要是由于分子内NH和OH的伸缩振动引起;在1646 cm-1、1556 cm-1和1235 cm-1存在振动吸收峰,这分别由明胶中的C=O(酰胺I带)伸缩振动、N-H(酰胺II带)变角振动、C-N(酰胺III带)伸缩振动所引起[39]。而SFE同样在3450 cm-1存在较宽的吸收峰,是由于其多糖中的O-H分子内伸缩振动引起;在2937 cm-1和1422 cm-1为水提物中C-H伸缩振动和变角振动引起的吸收峰;在1619 cm-1和1422 cm-1由羰基C=O和COO-伸缩振动引起的吸收峰;在1257 cm-1和815 cm-1存在由硫酸基S=O伸缩振动和C-O-S的不对称伸缩振动引起的吸收峰[40]。

图5 FG、SFE与不同比质量比的SFE-FG复聚物的红外光谱Fig.5 FT-IR spectra of FG, SFE, and SFE-FG coacervate prepared at different mass ratio
当形成SFE-FG复聚物时,O-H从3450 cm-1往3410 cm-1移动,发生往低波数方向移动现象。这与Zheng等[41]在研究明胶与结冷胶相互作用形成共混胶结果一致,推测可能是由于SFE与FG之间相互作用,非共价键引起复聚物链脱水。此外,复聚物的形成也使得FG的酰胺I带由1646 cm-1移动至1660 cm-1,酰胺II带从1556 cm-1移动至1548 cm-1。这与Bhowmik等[42]研究明胶与结晶纤维素互作的结果类似,推测可能是酯化作用引起键长变化而导致峰的移动。Guo等[43]研究豌豆分离蛋白(PPI)与果胶结合的复聚物,表明除氢键外,疏水和静电相互作用也参与了PPI-果胶复聚物的形成过程,而静电相互作用吸引力在形成PPI-果胶复聚物中起到更重要作用。
2.7 芯壁比对花色苷微胶囊包埋率的影响
芯壁比是通过影响芯材与壁材之间的疏水作用改变蛋白质的界面行为,进而影响复合凝聚的理化性质[22]。Ma等[44]发现适当的芯壁比有助于壁材高效率包埋,但是壁材比例过高会明显降低壁材的负载能力。因此,实验设置了不同的芯壁比,结果如图6可知,微胶囊的包埋率随着芯壁比的增加而变小,当芯壁比为2:8时,包埋率达到最大为91.84%。这与夏慧亭等[45]在包埋橄榄油的结果类似,推测可能是芯材含量的增加使未被包埋的芯材附着在微胶囊的表面,导致微胶囊粘连,囊壁破损引起芯材流失。如图7所示,当芯壁比为2:8、3:7时制备的微胶囊分散均一;而芯壁比为4:6时,微胶囊发生交联,微胶囊大小和形状改变,这可能是芯壁比过高导致的包埋率下降的原因。

图6 芯壁体积比对花色苷微胶囊包埋率的影响Fig.6 Effect of core/wall ratio (V/V) on the encapsulation efficiency anthocyanins microcapsules
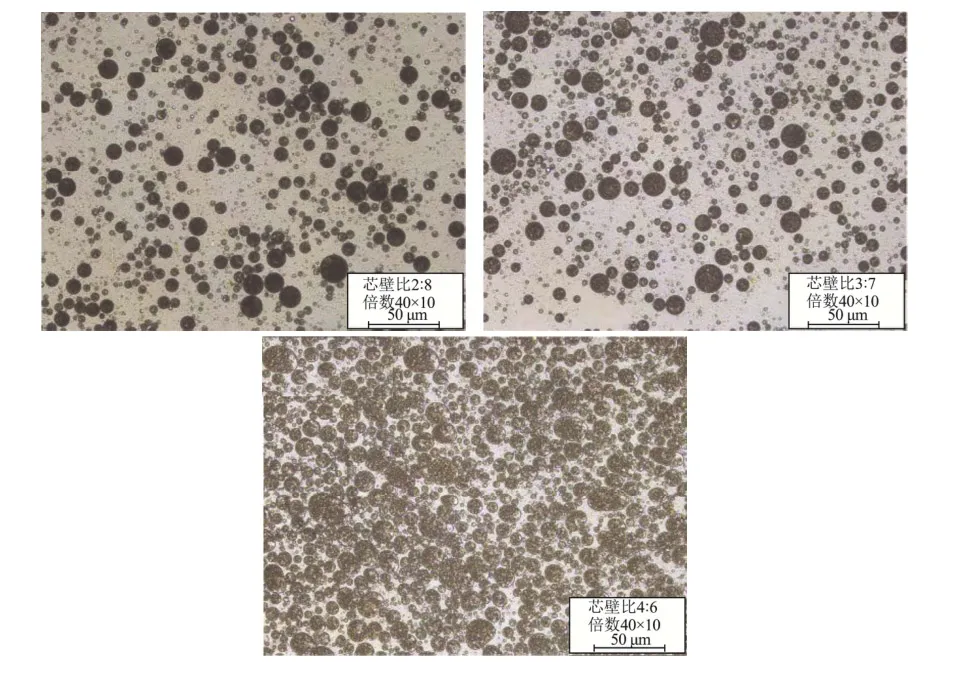
图7 芯壁体积比对花色苷微胶囊形态的影响Fig.7 Effect of core/wall ratio (V/V) on the morphology of anthocyanins microcapsules
2.8 壁材质量分数对花色苷微胶囊包埋率的影响
壁材质量分数也是影响微胶囊包埋率的重要指标之一,因此,在芯壁比为3:7,壁材SFE-FG复聚物比例为1:3,pH值4.2条件下,探究壁材质量分数对花色苷包埋率的影响。结果如图8所示,当壁材质量分数为0.5%和0.75%时,包埋率均低于60%。本结果与秦亚南等[46]研究壁材质量分数对葡萄籽油微胶囊的发现一致,可能是归因于壁材质量分数过低,进而引起复聚物产量不足,导致形成微胶囊的囊壁较薄,包埋不完全[47]。当壁材质量分数增加至1%时,包埋率可达到85%,值得注意的是这不仅归因于壁材质量分数的增加,而且可能与体系的粘度增加有关,据研究报道具有一定粘度的壁材溶液有利于微胶囊包埋[48]。另外,由于壁材质量分数超过1%时,SFE溶解性较差,且粘度过大,所以本实验未探究更高的壁材质量分数。由图9可知,微胶囊呈规则的球状,表面比较光滑。1%浓度的壁材相较于0.5%和0.75%浓度的壁材形成微胶囊粒径更小,微胶囊数量更多,壁材分子的利用率更高,进而使得微胶囊包埋率更高[49]。

图8 壁材质量分数对花色苷微胶囊包埋率的影响Fig.8 Effect of wall material concentration on the encapsulation efficiency anthocyanins microcapsules

图9 壁材质量分数对花色苷微胶囊形态的影响Fig.9 Effect of wall material concentration on the morphology of anthocyanins microcapsules
3 结论
本研究表明SFE是一种带负电的材料,且电荷量随pH降低而增多,可作为复合凝聚的天然负电材料。系统分析了SFE与FG复合凝聚反应过程,并确定了复合凝聚反应的适宜条件为SFE-FG复聚物质量比为1:3、pH值为4.2,此时SFE-FG复聚物产率为83.73%;在此条件下壁材质量分数1%、芯壁比为2:8时,具有较好的花色苷包埋率,高达91.84%。本研究首次采用SFE-FG复聚物作为包埋材料,且对花色苷的包埋率达到较高水平,表明SFE-FG复聚物在微胶囊应用上具有可行性和有效性,为天然提取物在复合凝聚微胶囊包埋技术的应用提供了重要的理论依据。
猜你喜欢
课堂内外(高中版)(2021年7期)2021-01-17
天津化工(2021年1期)2021-01-05
食品安全导刊·下旬刊(2020年3期)2020-07-09
中国测试(2018年3期)2018-05-14
中学生数理化·七年级数学人教版(2016年2期)2016-05-30
广东海洋大学学报(2015年3期)2015-12-22
中国塑料(2015年9期)2015-10-14
橡胶工业(2015年4期)2015-07-29
VOGUE服饰与美容(2015年6期)2015-05-30
水生生物学报(2015年1期)2015-02-28
