
同位素稀释-超高效液相色谱-串联质谱法测定鸡蛋中硝基咪唑类药物及其代谢物
2023-12-14 12:45:54邱肖依王冬梅李佳宽王梦颖管卓龙卢跃鹏涂凤琴
食品科学 2023年22期
邱肖依,王冬梅,李佳宽,许 晴,王梦颖,管卓龙,卢跃鹏,涂凤琴
(武汉食品化妆品检验所,国家市场监管重点实验室(食用油质量与安全),湖北 武汉 430040)
硝基咪唑类药物是一类含有5 位硝基取代咪唑杂环结构的化合物。甲硝唑(metronidazole,MNZ)、洛硝哒唑(ronidazole,RNZ)、地美硝唑(dimetridazole,DMZ)和异丙硝唑(ipronidazole,IPZ)是常见的硝基咪唑类药物,羟基甲硝唑(metronidazole-OH,MNZOH)、羟基异丙硝唑(ipronidazole-O H,IPZOH)和羟甲基甲硝咪唑(1-methyl-2-hydroxymethyl-5-nitroimidazole,HMMNI)分别是MNZ、IPZ、DMZ和RNZ的代谢物。硝基咪唑类药物被广泛用于治疗畜禽的滴虫病、厌氧菌感染等疾病[1],有不法商家将其添加到饲料中可提高饲料的转化率,增加动物的体质量。由于MNZ、RNZ、DMZ等含有硝基杂环的化合物具有潜在致癌、致畸作用[2],欧盟理事会将其列入A类禁用药物,不得在任何食品中检出[3-4],2002年我国农业部第193号公告和国家药品监督管理局第227号公告中规定,甲硝唑、地美硝唑及其盐、酯及制剂不允许以促进动物生长为目的在所有食品动物中使用。GB 31650—2019《食品中兽药最大残留限量》[5]明确规定地美硝唑、甲硝唑允许作治疗作用,但是不得在动物食品中检出。2019年我国农业农村部第250号公告明确规定食品动物中禁止使用洛硝达唑和替硝唑。鸡蛋是我国居民日常膳食中极为重要的营养食物,其质量安全事关人民群众身体健康。在日常监督抽检工作中发现,蛋鸡的养殖过程存在使用硝基咪唑类药物的情况,鸡蛋中检出硝基咪唑类药物的现象时有发生,因此有必要开发硝基咪唑类药物的快速检测方法,进一步提高鸡蛋质量安全风险监测水平。
目前报道的检测硝基咪唑类药物的测定方法主要有液相色谱法[6-10]、气相色谱-质谱联用法[11]、液相色谱-串联质谱法[2-3,12-19]。对比3 种检测方式,液相色谱法存在灵敏性不高、无法准确定性的缺点;气相色谱-质谱联用法前处理过程通常需要衍生化,且RNZ和HMMNI衍生产物相同,气相色谱-质谱联用法无法对两者区分;液相色谱-串联质谱法由于其灵敏度高、选择性好、无需衍生化,在复杂基质样品硝基咪唑类药物的分析检测中应用最为广泛。我国现行有效的动物性食品中硝基咪唑类药物的检测标准主要包括GB 31658.23—2022《动物性食品中硝基咪唑类药物残留量的测定 液相色谱-串联质谱法》[20]和GB/T 21318—2007《动物源食品中硝基咪唑残留量检验方法》[21],前者规定了对猪牛羊和鸡的肌肉和肝肾中MNZ、MNZOH、DMZ和HMMNI残留量进行测定,后者的适用范围包括猪鸡牛的肌肉和肝肾、鱼肉、奶粉和蜂蜜,两者的测定范围均不包含鸡蛋。鸡蛋中硝基咪唑类药物的检测方法主要参照SN/T 2624—2010《动物源性食品中多种碱性药物残留量的检测方法》[22],而该方法净化过程使用两个固相萃取柱,前处理操作较繁琐。当前,大部分文献采用固相萃取法对提取液进行富集和净化[6-9,12,15-16,23-24]。张丽媛等[15]建立液相色谱-串联质谱法测定鸡肉及鸡蛋样品中3 种硝基咪唑类药物,PCX固相萃取柱净化,定量限为5 μg/kg。Xia Xi等[23]采用SCX小柱对样品进行富集净化,结合超高效液相色谱-串联质谱法测定猪肾中硝基咪唑类药物及其代谢物。马东杰等[24]对比了PCX、MCX、SCX三种阳离子固相萃取柱对硝基咪唑类化合物的净化效果,发现DMZ的平均回收率在50%左右,IPZ的平均回收率小于10%,使用HLB净化IPZ的平均回收率也不到10%,使用PRiME HLB固相萃取柱MNZ、RNZ、IPZ、DMZ的平均回收率均达到70%以上。固相萃取柱的使用通常需活化、上样、淋洗、洗脱,过程耗时,净化过程中目标物流失、洗脱不完全等原因都会对实验的回收率产生不利影响,且使用成本高。近年来,QuEChERS作为一种快速、简单、廉价、高效、安全的前处理技术,在动物性食品兽药残留检测中应用较为广泛[10,14,25-26,29-31]。
本研究经过实验对比,采用十八烷基键合硅胶(C18)作为净化吸附剂,基于改良的QuEChERS技术结合同位素稀释-超高效液相色谱-串联质谱,建立了一种同时测定鸡蛋中硝基咪唑类药物及其代谢物残留的分析方法。该方法操作简便、定量结果准确、灵敏度高,使用弗帕斯检测实验室能力验证(food analysis performance assessment scheme,FAPAS)对样品进行验证,具有良好的准确度和精密度,适用于大批量样品的快速测定。
1 材料与方法
1.1 材料与试剂
硝基咪唑类化合物及内标(100 mg/L)(MNZ、RNZ、DMZ、IPZ、MNZOH、HMMNI、IPZOH、RNZ-D3、DMZ-D3、IPZ-D3、MNZOH-D2、HMMNI-D3、IPZOH-D3)北京曼哈格生物科技有限公司;乙腈(色谱纯)德国Merck公司;甲酸(色谱纯)上海阿拉丁生化科技股份有限公司;Oasis PRiME HLB固相萃取小柱(200 mg/6 mL)美国Waters公司;CNWBOND HC-C18SPE填料(40~63 μm)上海安谱科学仪器有限公司;水为Milli-Q系统制备的超纯水(18.2 MΩ·cm)。
2022年FAPAS鸡蛋中硝基咪唑类药物的测定能力验证样品(产品编号FCVD13-EGG2)由英国食品与环境研究院提供。
1.2 仪器与设备
Q-TRAP 6500质谱仪(配电喷雾电离Turbo V离子源)美国AB SCIEX公司;LC 30A液相色谱仪日本Shimadzu公司;Allegra X-15R离心机 美国Beckman公司;Vortex-Genie 2涡旋振荡仪 美国SCND公司;PM5-2000TL超声波清洗器 普律玛仪器公司;N-EVAP水浴氮吹仪 美国OA公司;ME204电子天平 瑞士梅特勒-托利多公司。
1.3 方法
1.3.1 色谱条件
色谱柱:Agilent Eclipse Plus C18柱(150 mm×2.1 mm,1.8 μm);柱温:40 ℃;进样量:2 μL,流动相:A为0.1%(体积分数)甲酸溶液,B为乙腈;流速:0.3 mL/min;梯度洗脱程序:0~1.0 min,90.0% A、10.0% B;1.0~3.0 min,90.0%~40.0% A、10.0%~60.0% B;3.0~3.5 min,40.0%~10.0% A、60.0%~90.0% B;3.50~6.01min,10.0% A、90.0% B;6.01~ 6.02 min,10.0%~90.0% A、90.0%~10.0% B;6.02~9.00 min,90.0% A、10.0% B。
1.3.2 质谱条件
离子源:电喷雾电离(electronspray ionization,ESI)正离子模式;检测方式:多反应监测(multiple reaction monitoring,MRM)模式;离子源温度500 ℃,电喷雾电压5 000 V;气帘气压力275.79 kPa;雾化气压力379.21 kPa;辅助加热气压力379.21 kPa;碰撞气体Medium。其他质谱参数见表1。
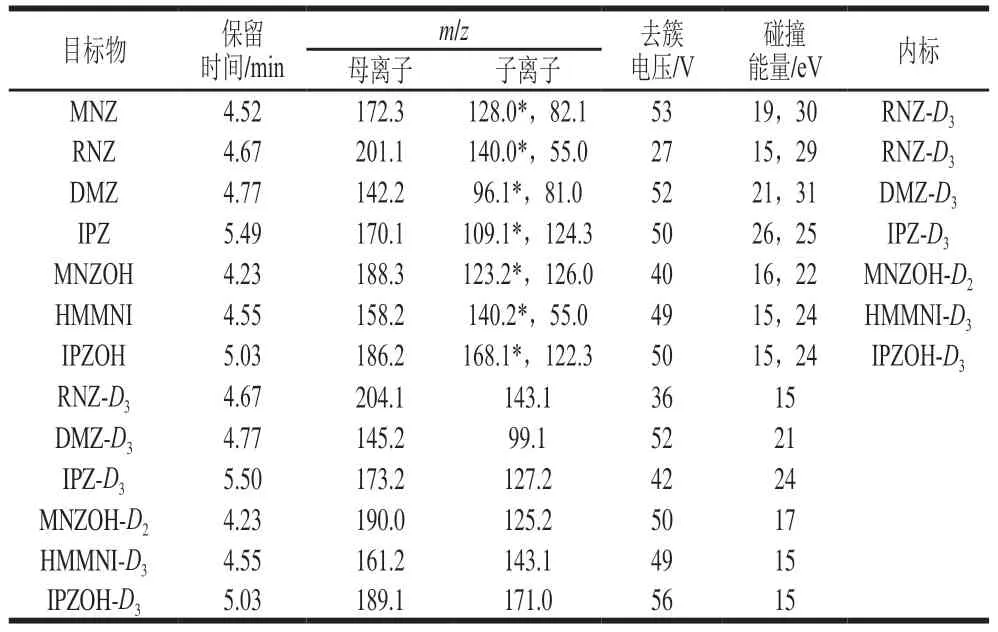
表1 硝基咪唑类药物及代谢物质谱采集参数Table 1 MS parameters for 5-nitroimidazoles and their metabolites
1.3.3 标准工作溶液的配制
标准溶液的制备:准确移取1 mL的RNZ、MNZ、IPZ、DMZ、MNZOH、HMMNI、IPZOH标准物质至10 mL棕色容量瓶中,用甲醇稀释,配制成10 mg/L的混合标准储备液。准确移取上述硝基咪唑类混合标准储备液1 mL至100 mL棕色容量瓶中,用甲醇稀释并定容至刻度,配制质量浓度为100 ng/mL的硝基咪唑类混合标准中间溶液。准确移取1 mL的RNZ-D3、DMZ-D3、IPZ-D3、MNZOH-D2、HMMNI-D3、IPZOH-D3标准物质,用甲醇溶解配制成10 mg/L的硝基咪唑类混合内标储备液。准确移取硝基咪唑类混合内标标准储备液1 mL至100 mL棕色容量瓶中,用甲醇稀释并定容至刻度,配制质量浓度为100 ng/mL的混合内标标准中间溶液。
取适量硝基咪唑类混合标准中间液、硝基咪唑类混合内标标准中间液,用体积分数10%乙腈溶液配制成0.5~20 ng/mL、内标质量浓度为5 ng/mL的工作曲线。
1.3.4 试样制备
新鲜鸡蛋洗净后去壳,用组织匀浆机搅拌均匀,置于聚乙烯瓶中,于-20 ℃储存。
1.3.5 提取
准确称取2.0 g(精确到0.01g)混匀后的样品,加入100 μL 100 ng/mL的硝基咪唑类混合内标标准中间溶液,再加入10 mL乙腈和1 g NaCl涡旋混合10 min,超声10 min,以4 500 r/min离心5 min,上清液待净化。
1.3.6 净化
取5.0 mL上述提取液,加入400 mg C18,涡旋2 min,以4 500 r/min离心5 min,取全部上清液加入2mL乙腈饱和正己烷,涡旋1 min,4 500 r/min离心5 min,弃去正己烷层,于40 ℃氮吹干,加入1.0 mL 10%(体积分数)乙腈溶液复溶,过0.22 μm有机滤膜,待测。
1.4 数据处理
采用Analyst软件(AB SCIEX公司)对研究的硝基咪唑类药物进行超高效液相色谱-串联质谱分析、数据采集和处理,采用Microsoft Excel进行统计计算和图表绘制。
2 结果与分析
2.1 仪器条件优化
2.1.1 质谱参数优化
使用流动注射泵以恒流方式进样,分别将质量浓度为100 ng/mL的7 种硝基咪唑类化合物的标准溶液,以及氘代内标标准溶液依次注入到离子源中,在ESI正离子模式下进行母离子全扫描,得到最优的各待测目标物的分子离子峰。以确定的分子离子峰为母离子进行二级质谱碎片离子扫描,获得碎片离子信息,得到二级质谱图,选取丰度较高子离子。根据前面选出的母、子离子,组建MRM离子对,优化碰撞能量与去簇电压并确定定量离子及定性离子。优化后相关参数见1.3.2节。选出的离子对数量符合EC/657指令中使用质谱法确证的4 个识别位点的要求。
2.1.2 色谱条件优化
目前报道的关于测定硝基咪唑类药物的文献[23-29]和标准[20-21],分析柱均使用反相色谱柱,增加色谱柱长能更有效地提高样品基质中目标分析物和共流出物的分离度并降低基质效应(matrix effect,ME)影响[27],因此本方法采用选用柱长150 mm的C18色谱柱进行分析。实验选用Waters XBridge C18(2.1 mm×150 mm,5 μm)和Agilent EclipsePlus C18(2.1 mm×150 mm,1.8 μm)两种色谱柱进行考察。结果表明超微填料(粒径≤2 μm)Agilent EclipsePlus C18色谱柱可以获得更好的分离效果,色谱峰峰形尖锐对称。流动相采用有机相+水相改善出峰情况,有机相比较乙腈和甲醇,两者都可获得较好的色谱峰形和信号响应,使用乙腈目标物能更好地实现基线分离,且可减少溶剂效应的干扰;在正离子扫描模式下,水相中甲酸的加入能够增加目标物响应,改善目标物的峰形,进一步比较0.1%的甲酸溶液和5 mmol/L乙酸铵溶液(含0.1%甲酸),结果表明乙酸铵加入后部分色谱峰响应降低。因此实验使用Agilent EclipsePlus C18色谱柱作为分析柱,选择0.1%的甲酸溶液-乙腈作为系统流动相。不同粒径填料的色谱柱及不同流动相对7 种硝基咪唑类化合物影响见图1。
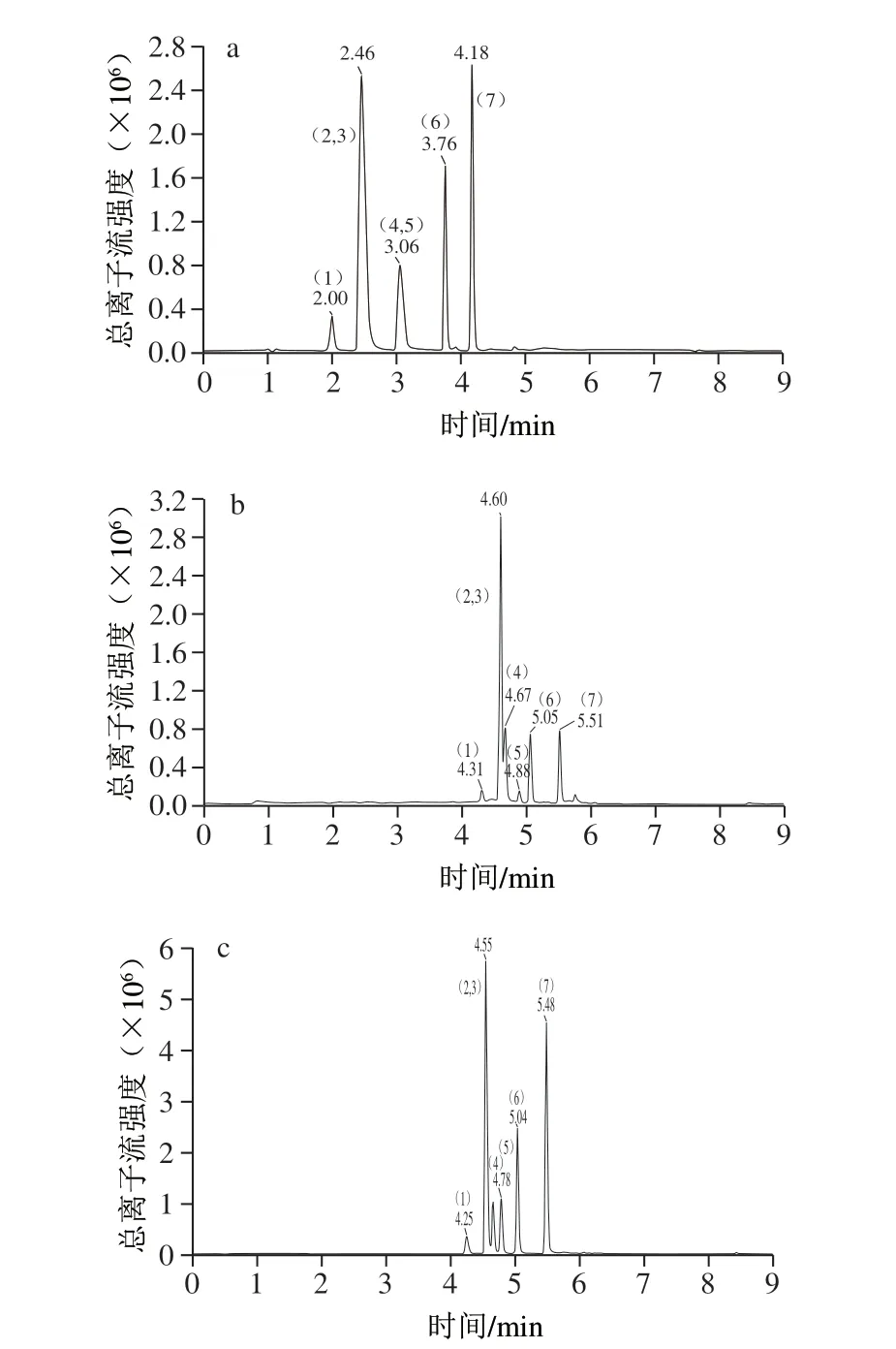
图1 7 种硝基咪唑类化合物的总离子流图Fig.1 Total ion current chromatograms of seven NMZs
2.2 提取溶剂的优化
在7 种硝基咪唑类化合物均未检出的空白鸡蛋样品中加入5 μg/kg硝基咪唑类化合物混合标准溶液,分别以乙腈、1%甲酸-乙腈、甲醇、乙酸乙酯有机溶剂作为提取液,对比7 种硝基咪唑类化合物的提取效果。结果表明:甲醇和乙酸乙酯的盐析效果较差,不利于分层,且甲醇提取液杂质较多,后续净化复杂,使用1%甲酸-乙腈和乙腈提取时,回收率无明显差异,乙腈具有沉淀蛋白等作用,提取液颜色较浅且各目标物回收率均满足实验要求,综合考虑,选择乙腈作为提取液。不同提取溶剂对硝基咪唑类化合物回收率的影响见图2。
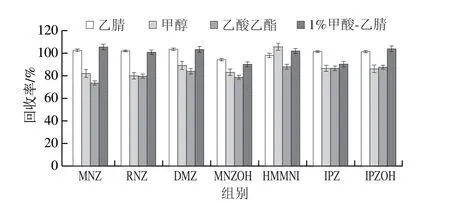
图2 不同提取溶剂对硝基咪唑类化合物回收率的影响Fig.2 Effects of different extraction solvents on the recoveries of NMZs
2.3 净化方式的优化
近年来,新型净化固相萃取小柱Prime HLB和QuEChERS作为快速高效的前处理技术,在动物性食品药物残留检测中应用较为广泛[10,14,24-26,28-31]。实验对比Prime HLB和C18对硝基咪唑类药物的净化处理效果,结果表明,Prime HLB和C18的平均回收率没有明显差异,不同净化材料对目标物回收率的影响如图3所示。Prime HLB采用亲水亲脂平衡填料,使用通过策略进行固相萃取净化,可获得相对较好的回收率,C18对于除去非极性杂质有较好的效果,部分化合物的回收率较采用Prime HLB稍低。但是鸡蛋基质复杂,采用Prime HLB净化后部分化合物(HMMNI和IPZOH)目标峰附近仍有干扰峰,影响定量结果准确性,所以选择C18为前处理净化方式。经过两种净化材料处理后HMMNI和IPZOH提取离子流图见图4。
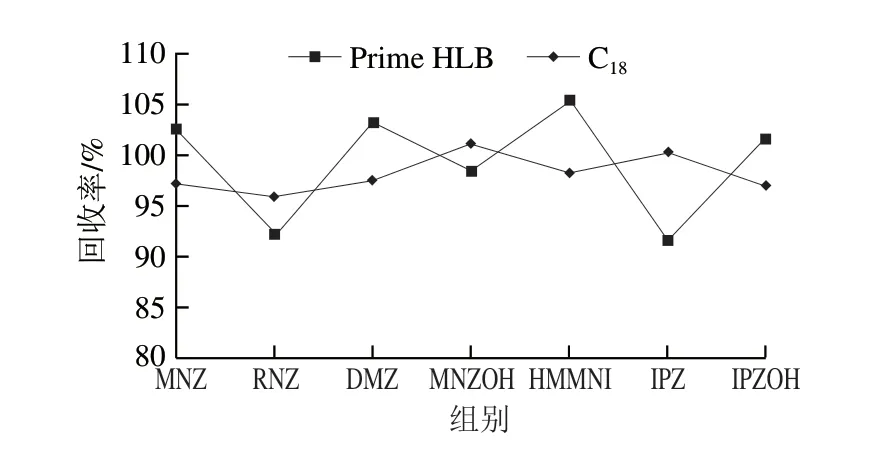
图3 两种净化方式的净化效率对比Fig.3 Effect of purification methods on the recoveries of NMZs
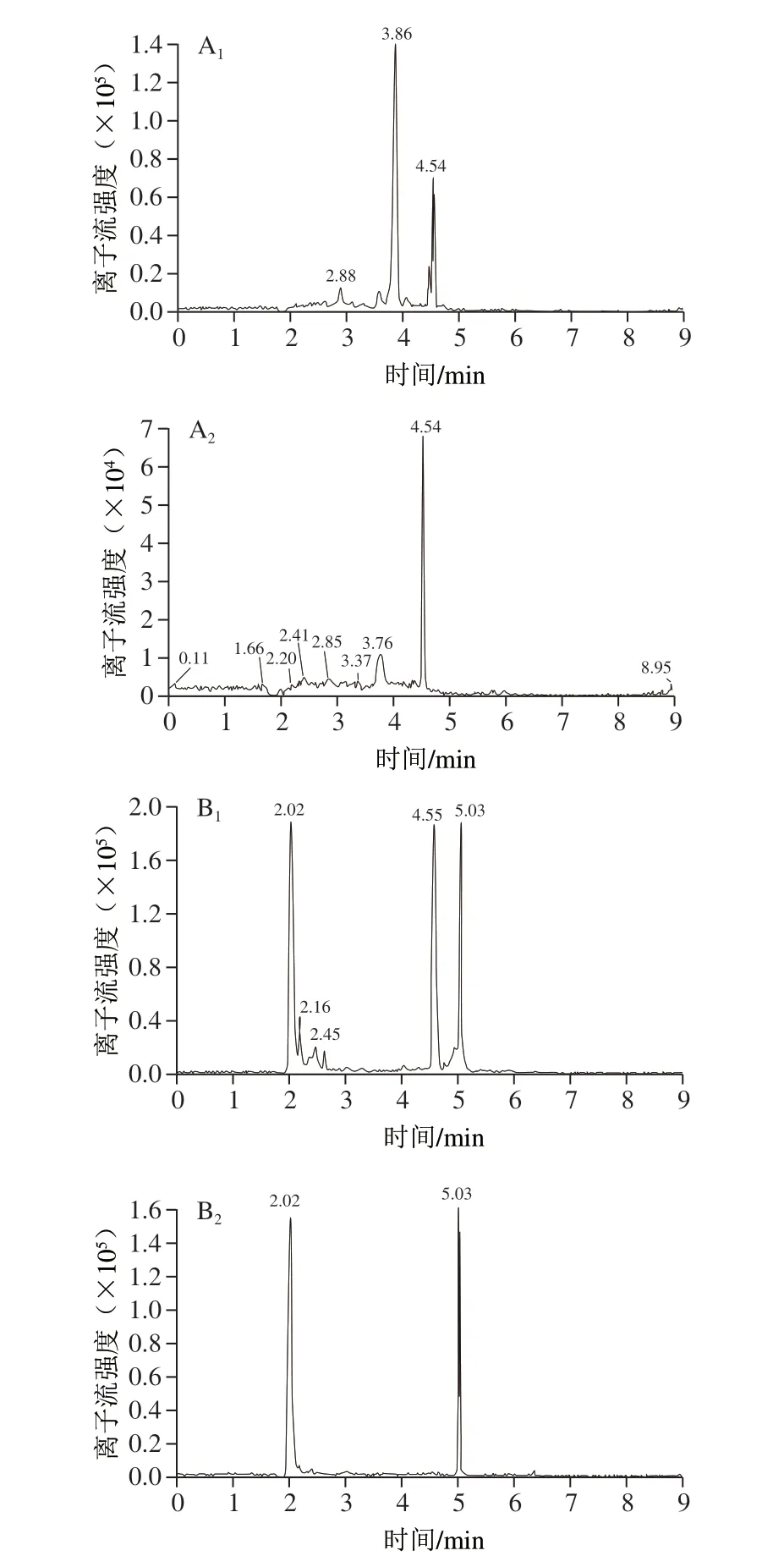
图4 HMMNI和IPZOH提取离子流图Fig.4 Extracted ion chromatograms of HMMN and IPZOH
实验进一步考察C18不同用量(50、200、400、600 mg)对目标化合物的影响。结果表明C18用量600 mg时DMZ和IPZ峰面积相对50 mg C18增加17.8%和31.8%,C18用量400 mg HMMNI峰面积相对50 mg C18增加了6.18%,MNZ、RNZ、MNZOH和IPZOH的峰面积有下降趋势,对比添加量50 mg和400 mg峰面积相对偏差为3.01%、3.70%、4.86%和6.39%,差异不明显,硝基咪唑类化合物的峰面积与C18用量关系见图5。使用400 mg C18填料对部分目标物存在吸附作用,但是吸附作用不明显,但是净化后HMMNI绝对响应峰面积增加,DMZ和IPZ绝对响应峰面积增加明显,综上分析,使用400 mg C18作为合适的吸附剂用量。
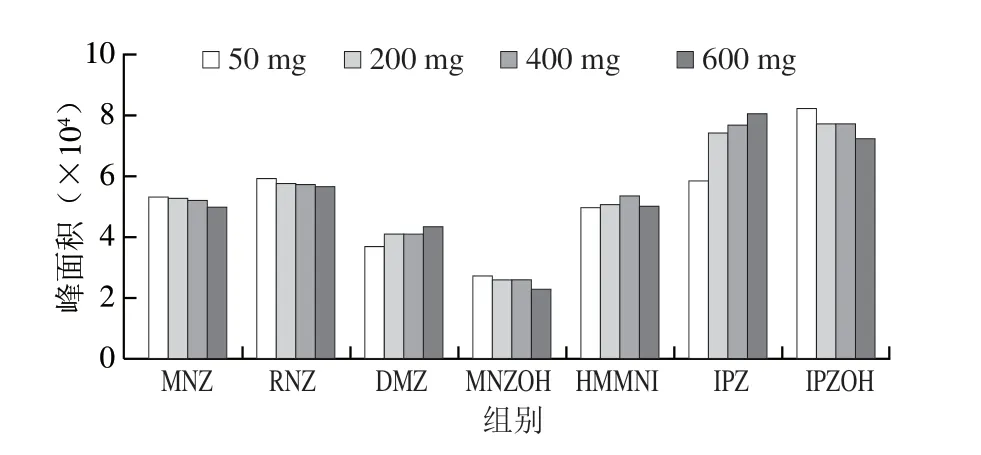
图5 C18吸附剂用量对硝基咪唑类化合物峰面积的影响Fig.5 Effects of the amount of C18 sorbents on the peak areas of NMZs
2.4 方法评价
2.4.1 线性范围、检出限(limit of detection,LOD)和定量限(limit of quantification,LOQ)将标准及内标混合系列工作液分别注入液相色谱-串联质谱仪中,测定相应的峰面积,以标准系列工作液中硝基咪唑化合物的质量浓度为横坐标,以硝基咪唑化合物与其内标的峰面积的比值为纵坐标,绘制标准曲线。结果表明,硝基咪唑化合物在0.5~20.0 ng/mL范围内线性相关系数(r)均大于0.999,呈现良好的线性关系(表2)。以信噪比为3确定方法LOD,以信噪比为10确定方法LOQ,得出鸡蛋基质中硝基咪唑及其代谢物的LOD在0.1~0.25 μg/kg之间,LOQ在0.3~1.0 μg/kg之间,方法的LOD均低于标准方法SN/T 2624—2010的检测低限(1 μg/kg),方法灵敏度较高。
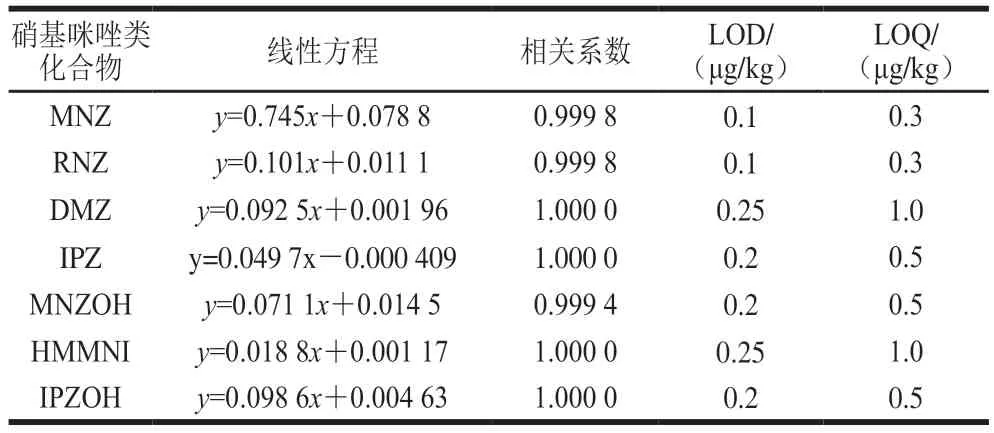
表2 硝基咪唑类化合物线性方程、相关系数、LOD和LOQTable 2 Linear equations,correlation coefficients,LODs,LOQs of NMZs
2.4.2 回收率与精密度
对空白样品进行低、中、高3 个水平的添加回收实验,硝基咪唑类化合物添加量分别为0.5、1.0、5.0 μg/kg,每个添加水平进行6 次平行实验,以考察方法的回收率和精密度。结果表明(表3),鸡蛋中7 种硝基咪唑类的回收率为96.5%~112.7%,相对标准偏差(relative standard deviation,RSD)为0.82%~8.14%。符合标准GB/T 27404—2008《实验室质量控制规范 食品理化检测》[32]规定的回收率和精密度的要求。
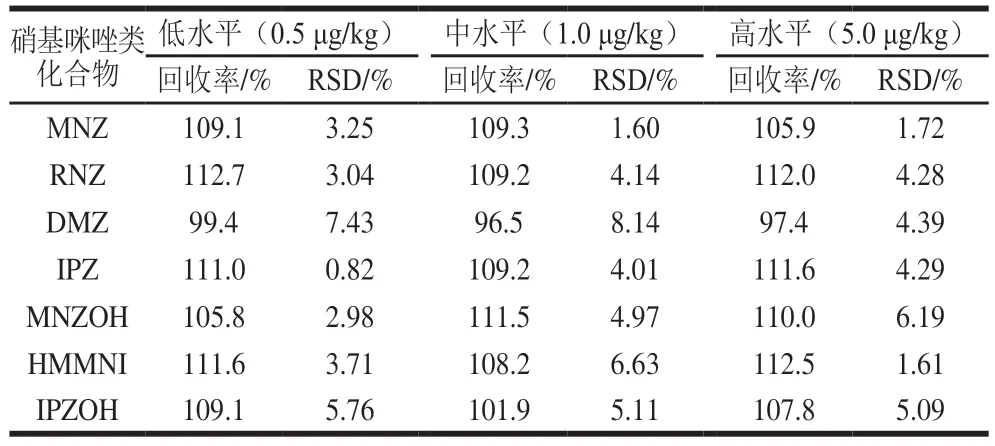
表3 硝基咪唑类化合物的加标回收率和精密度Table 3 Recoveries and precision RSDs of NMZs from spiked samples
2.4.3 ME的评价
方法按照(ME=基质标准曲线斜率/溶剂标准曲线斜率)评价ME。当ME小于0.85时,存在基质抑制效应;当ME为0.85~1.15时,ME不明显;当ME大于1.15时,存在基质增强效应[33]。准确称取2.0 g样品,加入10 mL乙腈和1 g NaCl旋涡混合10 min,超声 10 min,以4 500 r/min离心5 min,取5.0 mL上清液,净化处理后加入7 种硝基咪唑类化合物和内标标准溶液,配制成0.5~20 ng/mL基质标准工作溶液。分别作基质和溶剂的外标和内标曲线,内标质量浓度为5 ng/mL。结果表明,除HMMNI(MEex=0.774)和RNZ(MEex=0.814)存在抑制效应外,其他几种硝基咪唑类化合物经过本法净化后各基质溶液中ME不明显。方法通过内标法进行矫正后,7 种硝基咪唑类化合物的ME不明显,因此本法采用溶剂标准曲线和内标法测定7 种硝基咪唑类化合物含量,ME评价如表4所示。
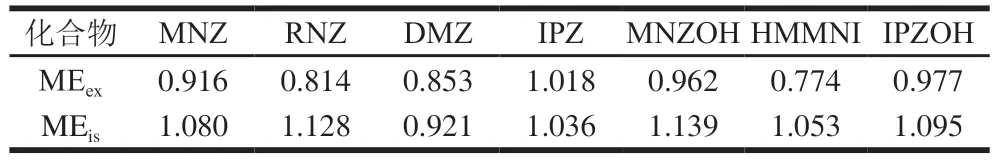
表4 硝基咪唑类化合物的ME评价Table 4 Matrix effects of MNZs
2.4.4 方法比对与实际样品分析
采用建立的同位素稀释-超高效液相色谱-串联质谱法对FAPAS考核样品(编号02477)鸡蛋中的硝基咪唑类化合物进行测定,与标准方法SN/T 2624—2010结果进行比对。考核样待测目标物为本方法测定的7 种硝基咪唑类化合物,DMZ靶值为5.15 μg/kg,HMMNI 靶值为11.0 μg/kg,MNZ靶值为7.81 μg/kg,MNZOH靶值为5.17 μg/kg,RNZ、IPZ和IPZOH未检出,z值表示测定值与真值之间的偏离程度,|z|≤2则结果满意。未检出目标物采用加标回收的方式进行评价(加标量5.0 μg/kg)。两种方法对DMZ、HMMNI、MNZ、MNZOH、RNZ、IPZOH的测定结果(表5)比较接近,相对偏差分别为2.28%、3.36%、2.83%、6.82%、0.20%、9.90%。能力验证结果表明,本方法对鸡蛋中4 种硝基咪唑类化合物的测定结果均在|z|≤2的置信区间。RNZ、IPZ、IPZOH采用本方法测定的加标回收率分别为101%、106%和102%,SN/T 2624—2010的加标回收率分别为101%、76.6%和92.2%。综上,采用本方法对鸡蛋中7 种硝基咪唑类化合物测定结果满意。
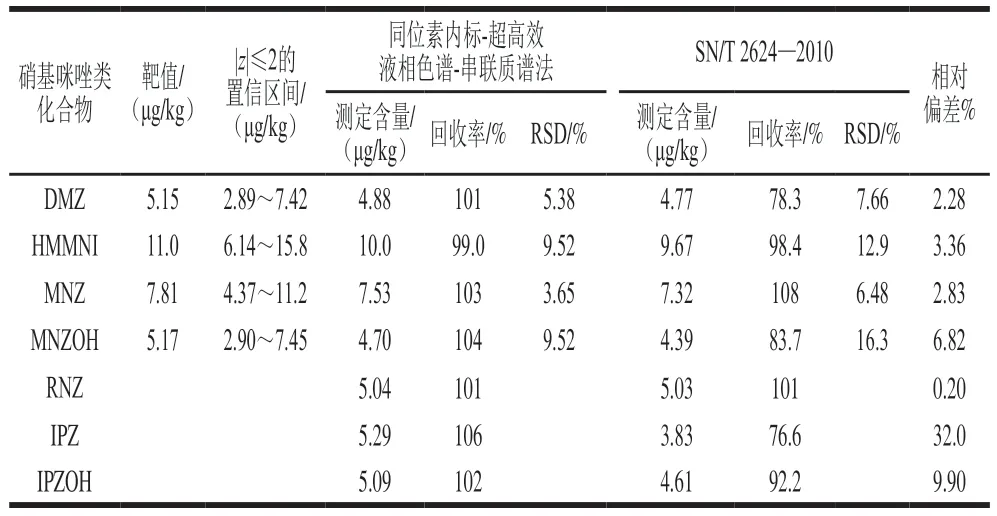
表5 鸡蛋中硝基咪唑类化合物的不同分析方法比对Table 5 Comparison of different analytic methods for detection of MNZs in eggs
应用建立的同位素稀释-超高效液相色谱-串联质谱法对购自超市和农产品批发市场的38 批次鸡蛋样品中7 种硝基咪唑类化合物进行测定。38 批次样品中,检出1 批次MNZ阳性样品,甲硝唑含量为320 μg/kg,超过限量1.0 μg/kg,其他样品均未检出7 种硝基咪唑类化合物,说明在国家明令禁止的情况下,仍存在不法分子违规使用硝基咪唑类药物的情况。
3 结论
本实验建立了同位素稀释-超高效液相色谱-串联质谱法同时测定鸡蛋中7 种硝基咪唑类药物的方法。方法LOD为0.1~0.25 μg/kg,LOQ为0.3~1.0 μg/kg,结果准确度高,前处理操作简便,具有快速、高效、易推广的优点,适用于大批量鸡蛋中硝基咪唑类药物的快速筛查、定性定量检测,对于鸡蛋中硝基咪唑类药物残留情况的监测具有重要的应用价值。
猜你喜欢
中学生学习报(2021年20期)2021-11-24 15:30:52
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
鞍钢技术(2021年3期)2021-06-11 05:13:50
化工管理(2021年7期)2021-05-13 00:46:24
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中成药(2018年6期)2018-07-11 03:01:32
中国农资(2016年1期)2016-12-01 05:21:23
中国粮油学报(2016年5期)2016-01-23 02:45:06
分析测试学报(2015年8期)2016-01-13 06:19:31
化工进展(2015年3期)2015-11-11 09:08:25
