
乙撑基对分子的构象、电导性质的影响
2023-12-06 06:29陈浩兵肖博怀周疆豪王瑞霞李云川
武汉科技大学学报 2023年6期
陈浩兵,王 旭,肖博怀,周疆豪,王瑞霞,李云川
(武汉科技大学材料学部,湖北 武汉,430081)
分子的构象是影响分子力学行为、溶解度和光电特性的重要因素。Qin等[1]报道了一种具有高平面性的共轭聚合物,将其用于太阳能电池可以实现5.4%的光电转化效率(PCE)。Jiang等[2]在合成非富勒烯受体ITMIC和ITCIC的基础上,通过研究发现,含S…Cl 非共价键构象锁(NCLs)的ITCIC分子平面性优于不含NCLs的ITMIC分子,将前者用于太阳能电池时相应的PCE也提高了近1倍。目前,已有的研究普遍认为,提高分子平面性可以降低分子在薄膜状态下的重组能,改善固态分子间的堆积状态,从而使得分子之间的电子耦合更强,更有利于电荷的长距离迁移[3-4]。因此,获得高平面性分子构象是一种设计高电导率材料的有效途径。
分子稠环化是改善分子平面构象、提高材料载流子迁移率的常用措施,但其存在单体合成复杂、目标产物溶解性差等不足[5-6]。相比之下, NCLs利用O…S、N…S、X…S(X=Cl、Br、F)等代表性原子间的相互作用来增强共轭骨架的平面性[7-8],具有合成简单、不影响材料溶解度等优点,是一种更理想的构建高平面性π-共轭分子的策略[9-10]。另外,朱生勃[11]的研究发现,利用乙撑基可以在不降低材料溶解度的前提下对有机半导体分子构象进行调控,这对设计高性能的有机半导体具有重要的借鉴意义。为了进一步在分子层面揭示乙撑基在电荷传输过程中的作用,本研究首先利用铃木偶联反应合成TBCN和Et-TBCN两种四联苯衍生物分子,再采用扫描隧道显微镜固结法(STM-FJ)研究这两种分子的单分子电导,基于四联苯分子骨架的构型及氰基锚定基团-电极耦合,探讨了乙撑基对分子电导的影响机制,以期为设计和开发高导电性有机半导体材料提供参考。
1 实验部分
1.1 材料合成
利用铃木偶联反应合成TBCN和Et-TBCN两种分子,合成路线见图1。在合成TBCN时,首先将4,4′-二溴-1,1′-联苯(400 mg)、4-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)苯腈(705 mg)、K2CO3(3.2 g)及H2O(12 mL)加入三口反应瓶中,再加入乙醇(40 mL)和甲苯(60 mL),在N2气氛下搅拌10 min后加入Pd(PPh3)4(58mg),继续通N2搅拌30 min,再加热至78 ℃并反应回流12 h。将所得混合物倒入冷水中,利用二氯甲烷(DCM)萃取,收集二氯甲烷层后借助无水硫酸钠干燥,再经减压蒸馏除去有机溶剂,使用色谱柱纯化粗产物,即可得到白色固体TBCN(246 mg,产率54%)。Et-TBCN的合成路线与TBCN基本相同,只是将4,4′-二溴-1,1′-联苯替换为2,7-二溴-9,10-二氢菲,最后得到白色固体Et-TBCN(127 mg,产率55%)。
1.2 样品制备
利用电化学方法腐蚀纯度为99.99%、直径为0.2 mm的金丝制得纳米金探针,腐蚀液为HCl和无水乙醇按体积比1∶1配置。接着,依次用食人鱼溶液(H2SO4、H2O2体积比为3∶1)、超纯水以及无水乙醇清洗探针,除去探针表面残留的有机物杂质。用于分子修饰的金基底采用氢火焰退火,从而获得具有(111)晶面的平整金片。
1.3 STM-FJ法原理及测试方法
采用STM-FJ法测量分子的电导,测试原理见图2。通过控制金纳米探针与金基底之间的间隙,将分子连接到间隙中形成金属/分子/金属结。当分子未连接或者从电极上离开时,观测到的电流信号为基线电流信号。当有单分子结形成时,基线电流上可能产生特征电流跳跃信号,通过统计即可获得单分子的电导数据。测试环境为1,2,4-三氯苯 (TCB)溶液,浓度为1 mmol/L,针尖偏压为0.1 V。
1.4 分子模拟方法
基于密度泛函理论(DFT)进行量子化学计算,在B3LYP/6-311G(d.p)基组水平下对TBCN和Et-TBCN分子进行几何优化,所有优化均使用 Gaussian 09 软件包在298 K下进行。
2 结果与讨论
2.1 单分子的电导分析
图3所示为TBCN、Et-TBCN分子以及无目标分子的对照组样品的典型电流-时间(I-t)曲线。由图3可见,对照组样品的I-t曲线上观测不到特征电流的跳跃,只显示背景电流噪音;而加入目标分子TBCN或者Et-TBCN后,相应的I-t曲线上出现明显的特征电流跳跃,这些电流跳跃信号来源于金电极之间形成的分子结。在成功检测到目标分子特质电流信号的基础上,采集0.001、0.005、0.01、0.05、0.1、0.5、1 nA等不同基线电流下的样品I-t曲线,用以确定检测分子电导的最佳实验参数。根据测试结果,得到TBCN和Et-TBCN分子的成结情况以及目标分子的电导直方图如图4所示。由图4(a)可见,当基线电流为0.05 nA时,TBCN分子I-t曲线上出现的特征电流信号最多,此时的纳米电子间隙最有利于TBCN分子形成稳定的分子结。图4(b)所示的不同基线电流下TBCN分子的电导值统计结果显示,TBCN分子在基线电流分别为0.01、0.05 nA时的电导值接近,并且当基线电流为0.05 nA时,TBCN分子电导直方图的拟合度最高。相比之下,在其它基线电流下,TBCN分子I-t曲线上的特征电流信号较少且分散。这进一步证实测量TBCN分子电导的最佳基线电流为0.05 nA。通过分析图4(c)和图4(d)发现,测量Et-TBCN分子电导的最佳基线电流也是0.05 nA。这是因为TBCN和Et-TBCN分子长度相同,均为2.09 nm,故二者倾向于在同样大小的纳米电极间隙下形成分子结。基于上述测试结果,通过高斯拟合可确定TBCN和Et-TBCN分子的电导值分别为(1.74±0.23)×10-6G0和(3.09±0.17)×10-6G0,二者电导与四联苯衍生结构的电导[12]处在同一个数量级,表明该测量结果真实可靠。
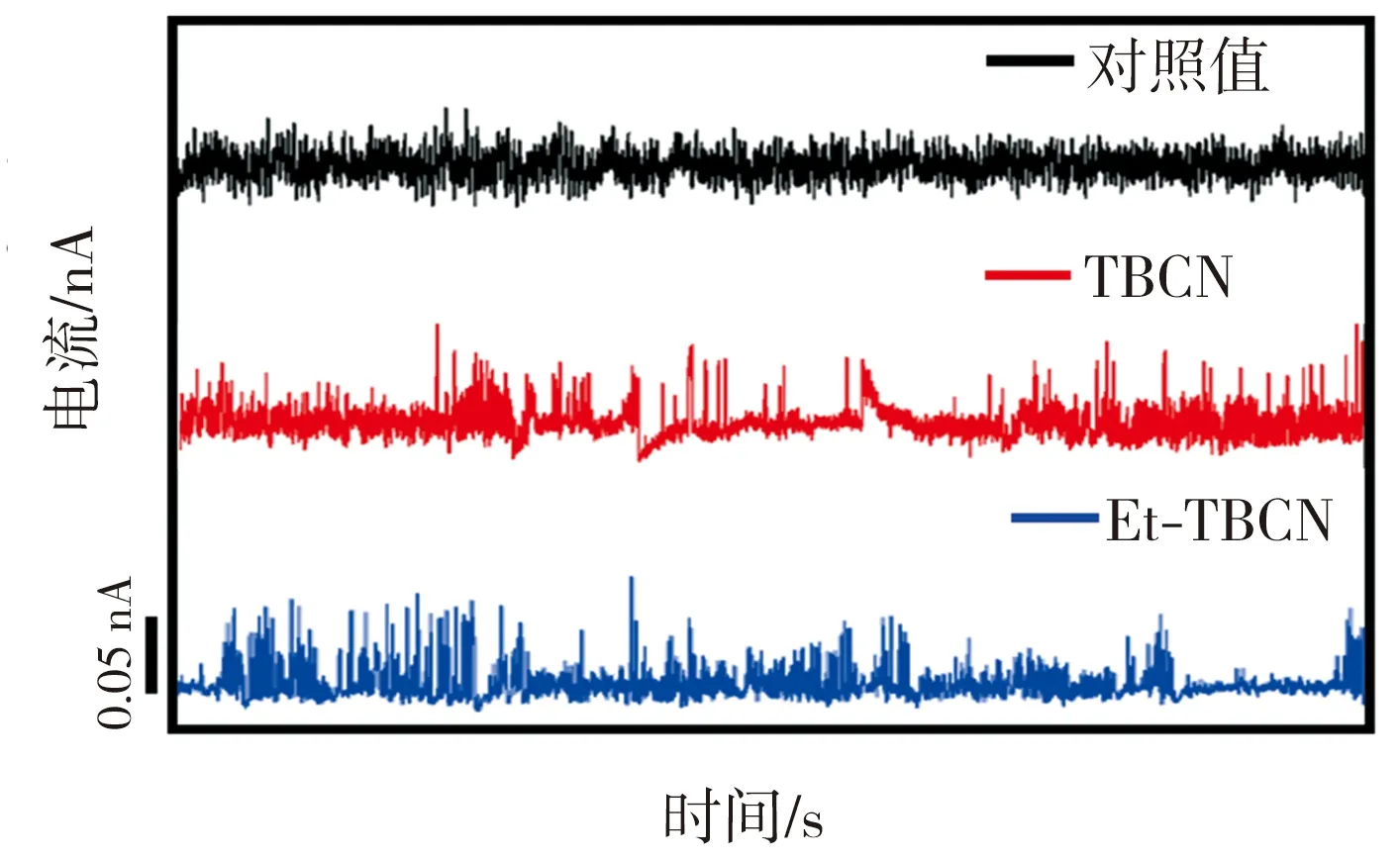
图3 样品的典型I-t曲线
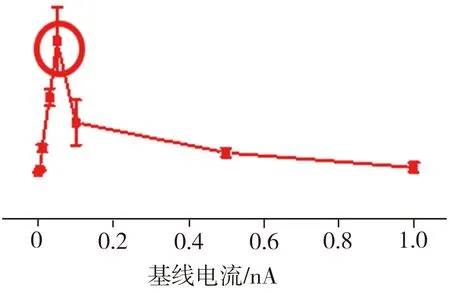
(a) TBCN分子成结率
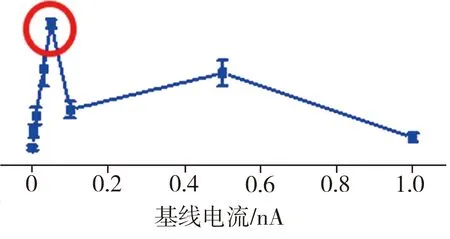
(c) Et-TBCN分子成结率
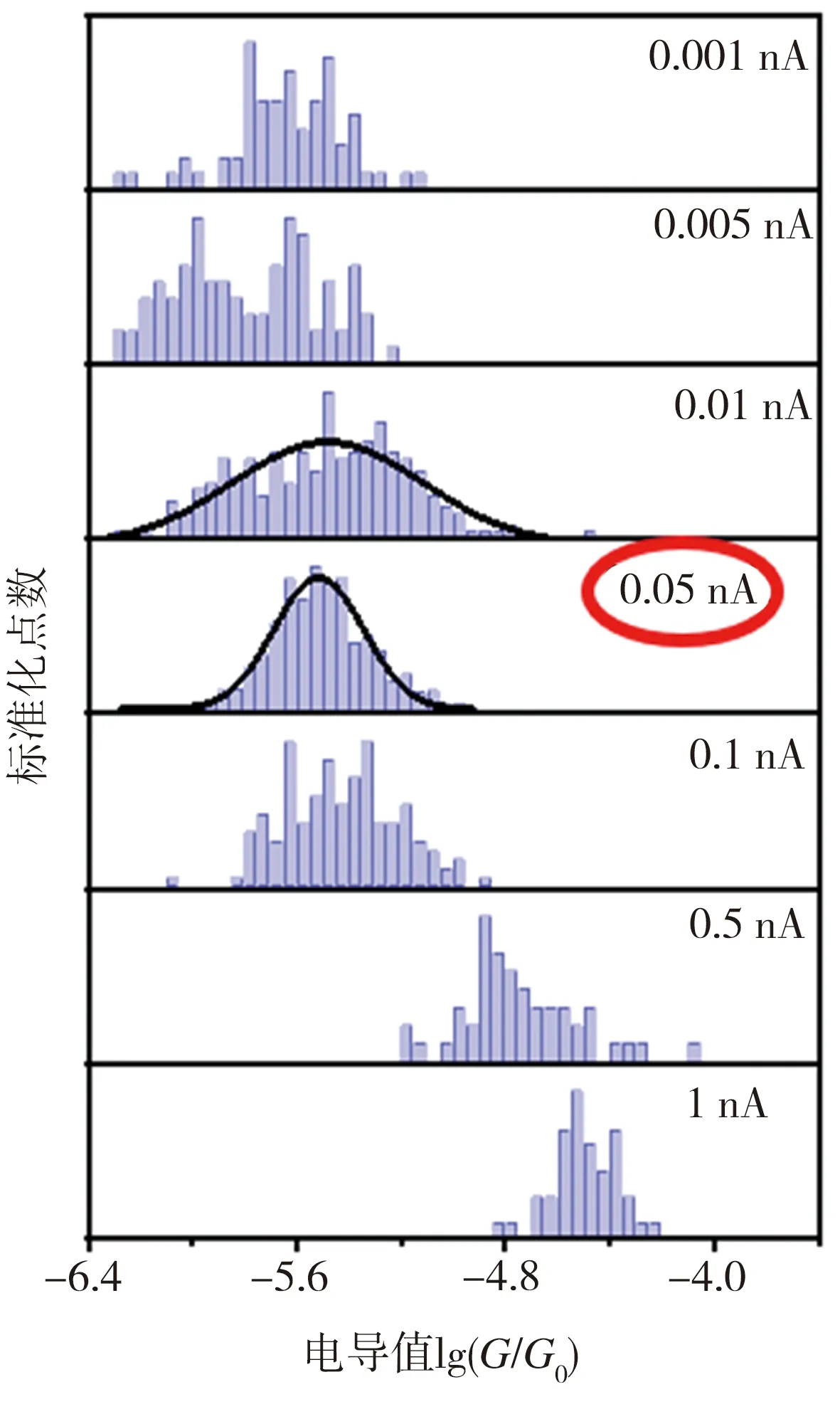
(d) Et-TBCN电导直方图
2.2 乙撑基对分子构象、电导的影响分析
经高斯拟合所得Et-TBCN分子电导较TBCN分子相应值增加了78%,表明乙撑基能显著地提升分子的电荷传输能力。对于共轭联苯分子,穿过分子的隧穿电流与分子的平面构象密切相关[13]。基于此,本研究利用DFT计算方法对TBCN和Et-TBCN分子进行结构优化,并测定二者的二面角,结果如图5所示。由图5可见,TBCN分子四联苯骨架中的二面角θ1与Et-TBCN分子四联苯骨架中的二面角θ2存在较大差异,相应值分别为38.6°、20.8°,这表明乙撑基的引入增强了Et-TBCN分子的平面性。文献[14]指出,分子电导G与其骨架中二面角余弦值的平方存在正比关系。经计算,本研究中cos2θ2值是cos2θ1值的1.43倍,从理论上来说,因二面角减小的缘故,Et-TBCN分子的电导较TBCN分子相应值可提升43%。然而该增幅仍低于实测值78%,表明除了分子平面化之外,还存在其它提高分子电导的途径。
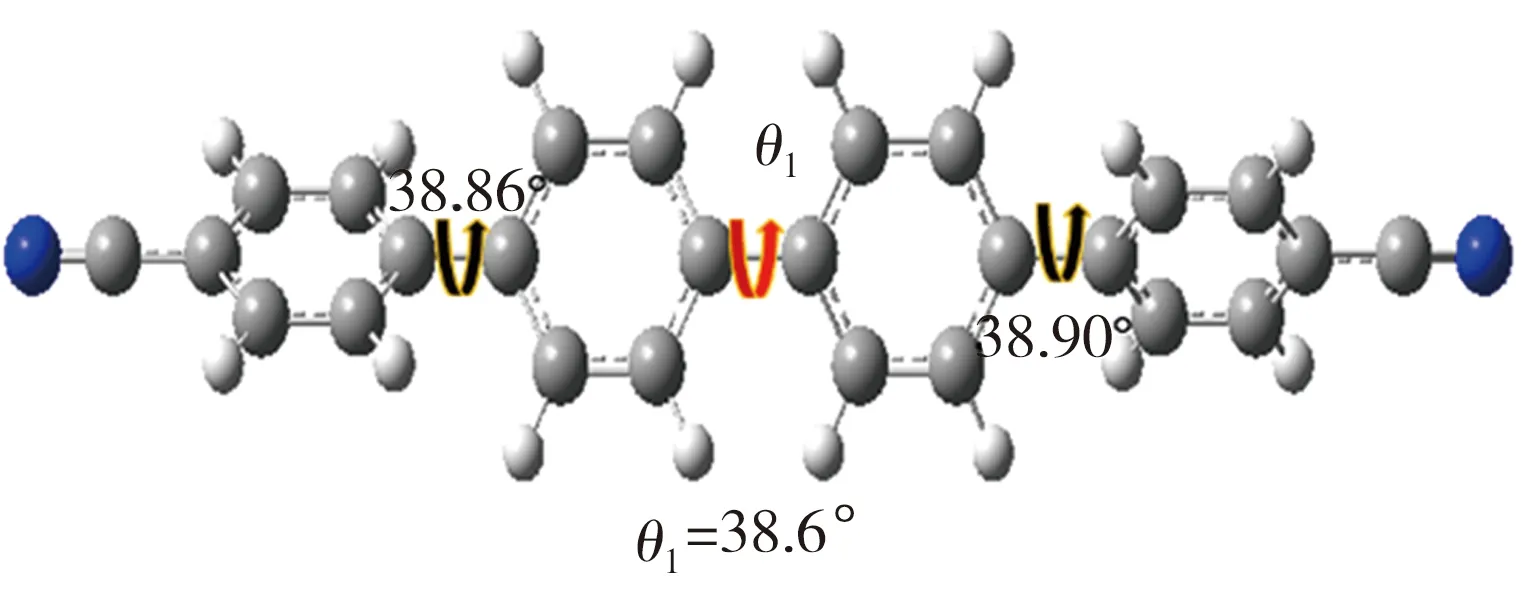
(a) TBCN优化结构及二面角
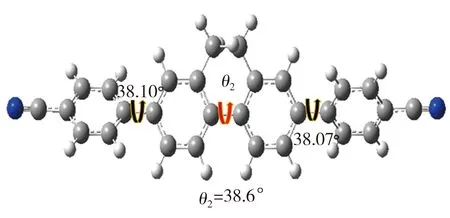
(b) Et-TBCN优化结构及二面角
2.3 乙撑基对分子-电极耦合、电导的影响分析
除了分子骨架,锚基-电极界面耦合也会影响分子的电导,这与分子能级HOMO/LUMO与金费米能级的相对位置有关。图6所示为TBCN和Et-TBCN分子的循环伏安、紫外吸收光谱测试结果以及能级分布情况。由循环伏安测试结果(图6(a))可以确定TBCN和Et-TBCN的氧化起始电位分别为1.83、1.74 eV[15],通过进一步计算可得二者的HOMO能级EHOMO分别为-6.10、-6.01 eV。计算公式[16]为:
EHOMO=-e[Eonset,ox+4.8-EFC]
(1)
式中:Eonset,ox为氧化起始位点,EFC是用于校正的二茂铁氧化还原电位0.53 eV。根据样品的紫外吸收光谱(图6(b)),可推导出TBCN和Et-TBCN的光学带隙(Eg)分别为-3.51、-3.33 eV,再结合HOMO能级和能级间隙可推算出TBCN和Et-TBCN的LUMO能级分别为-2.59、-2.68 eV(图6(c))。将目标分子的HOMO、LUMO能级与金电极的费米能级(-4.2 eV)[17]比较,发现其LUMO能级与金电极的费米能级更接近,这与文献[18]报道的以氰基为锚定基团的分子借助LUMO能级传输电荷的结论一致。因此,可以确定LUMO轨道是TBCN和Et-TBCN分子的主要电荷传输通道。此外,Et-TBCN分子的LUMO能级和金费米能极之间的隧穿势垒(ε)为1.52 eV,小于TBCN分子LUMO能级与后者之间的隧穿势垒(1.61 eV),这表明Et-TBCN分子能与金费米能级产生更强的电子耦合(Γ)。根据单洛伦兹电荷传导模型相关公式[19]:
G=G0[Γ2(ε2+Γ2)-1]
(2)
隧穿能垒更低、分子-电极耦合更强的Et-TBCN分子更易产生高电导,即乙撑基可以通过调整分子能级、减小分子带隙来促进电荷传输。
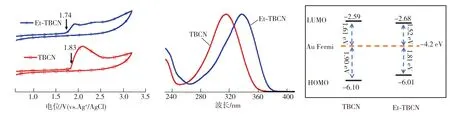
(a) C-V曲线 (b) 紫外吸收光谱 (c) 能级分布图
3 结语
本文通过单分子电导测量技术和理论模拟研究了乙撑基对四联苯衍生物的单分子电学特性的影响。实验结果表明,引入乙撑基使分子内的电荷传输效率增强。分子构型模拟表明乙撑基的引入提升了联苯的平面性,而分子能级研究表明乙撑基降低了分子的带隙。根据电导隧穿理论,这两个方面均对电导提升起正向作用。本研究在单分子层面揭示了乙撑基对于分子结电荷传输的作用,这将为设计和开发高导电性的有机半导体材料提供一定的实验和理论支撑。
猜你喜欢
中学生数理化(高中版.高考数学)(2022年2期)2022-04-26
新世纪智能(数学备考)(2021年4期)2021-08-06
中学生数理化(高中版.高考数学)(2020年3期)2020-05-25
高中生·天天向上(2018年1期)2018-04-14
温州大学学报(自然科学版)(2016年1期)2016-10-27
癌变·畸变·突变(2016年3期)2016-02-27
合成化学(2015年2期)2016-01-17
化工进展(2015年3期)2015-11-11
化学分析计量(2015年4期)2015-03-23
应用化工(2014年7期)2014-08-09
