
超声诊断儿童内脏反位伴胆道闭锁多脾综合征1例
2023-12-05 16:04祁平安
临床超声医学杂志 2023年11期
祁平安
患儿男,出生38 d,因重症肺炎、黄疸、大便呈陶土色来我院就诊。体格检查:全身皮肤黏膜黄染,右侧腹部较软,未触及肝脏。实验室检查:总胆红素252.3 μmol/L,直接胆红素192.2 μmol/L,总胆汁酸68.39 μmol/L。临床考虑胆汁淤积性肝炎。超声心动图检查:心脏大部分位于右侧胸腔,心尖指向右侧,心房反位,心室左襻,房室连接一致,房间隔缺损0.36 cm,降主动脉左侧可见奇静脉扩张并汇入上腔静脉。超声心动图提示:镜面右位心伴房间隔缺损,奇静脉增宽。腹部超声检查:左季肋区探及肝脏,右季肋区探及5个脾脏(图1A),胃位于右侧腹,降主动脉位于脊柱右前方,奇静脉位于降主动脉左后方(图1B),降主动脉前方未见下腔静脉汇入右房,降主动脉左后方可见扩张的奇静脉,三支肝静脉直接汇入右房。胆囊大小约1.48 cm×0.26 cm,胆囊内壁僵硬,餐后30 min胆囊未见明显收缩,大小、形态与餐前相同(图1C、D),门静脉内径0.41 cm,肝动脉内径0.21 cm,肝脏实质回声增粗增强,肝门部可见增厚的纤维层,厚约0.42 cm(图1E),肝脏包膜尚光滑,包膜下可见稀疏血流信号,肝门部未见胆管回声,肝内胆管未扩张,腹腔内可见游离液体,最深约1.50 cm。超声提示:胆道闭锁合并内脏反位伴多脾,结合超声心动图表现,考虑为左房异构综合征伴先天性胆道闭锁。CTA检查:内脏反位,多脾,肝下段下腔静脉缺如(图2)。临床以可疑胆道闭锁行腹腔镜检查,镜下见5 个脾脏位于右季肋区,肝脏位于左季肋区,肝脏增大呈暗红色,肝脏表面散在蜘蛛痣样血管,胆囊穿刺置管造影,未见造影剂向下显影,证实为胆道闭锁,患儿遂行开腹肝门-空肠吻合术。术中病理活检诊断:胆囊黏膜缺如,肝细胞肿胀,胆汁颗粒淤积,胆管板发育紊乱,细胆管增生,肝纤维化3 级,符合先天性胆道闭锁(图3)。术后1、3、6、9、12 个月复查超声提示肝脏回声稍增强,门静脉未见明显增宽,肝内胆管未扩张。实验室检查:胆红素明显下降,肝功能其他指标趋于正常。
讨论:左房异构综合征又称多脾综合征,是内脏异构综合征的一种,常合并复杂的先天性心脏畸形[1],合并心外畸形主要有多脾、肠旋转不良、双侧二叶肺、脐膨出、下消化道闭锁、尾退化不良、双肾发育不全、胆道闭锁等[2]。胆道闭锁多脾综合征(biliary atresia splenic malformation syndrome,BASM)最早由Davenport 等提出,以区分其他病因引起的胆道闭锁。BASM 诊断标准为内脏反位、多脾及胆道闭锁,典型超声表现为:①内脏反位:肝脏大部分位于左季肋区,脾脏大部分位于右季肋区,胃位于右季肋区;②多脾:右侧季肋区可见多个脾脏回声影,多呈月牙形,少数可呈圆形,类似于副脾;③胆道闭锁[3]:胆囊回声的异常,胆囊长径<1.5 cm 或呈条索状不充盈,胆囊壁僵硬,黏膜层显示不清,餐后胆囊无明显减小;因肝脏功能减低,肝动脉常代偿性增宽,内径多≥0.2 cm,流速加快;如肝脏发生胆汁淤积性肝纤维化,可表现为肝门部汇管区增厚的条索状高回声,厚度≥0.3 cm;此外,胆道闭锁还会继发肝脏包膜下血管增生,表现为肝脏包膜下血流信号增多。本例患儿超声表现为内脏反位、多脾及胆道闭锁,符合BASM的诊断。

图1 本例患儿上腹部超声图像(STOMAC:胃;LIVER:肝;DAO:降主动脉;AZV:奇静脉)
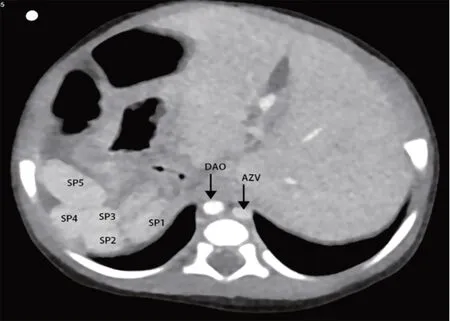
图2 本例患儿上腹部CTA 图示内脏反位,多脾,肝下段下腔静脉缺如(DAO:降主动脉;AZV:奇静脉;SP1~SP5:脾脏1~5)
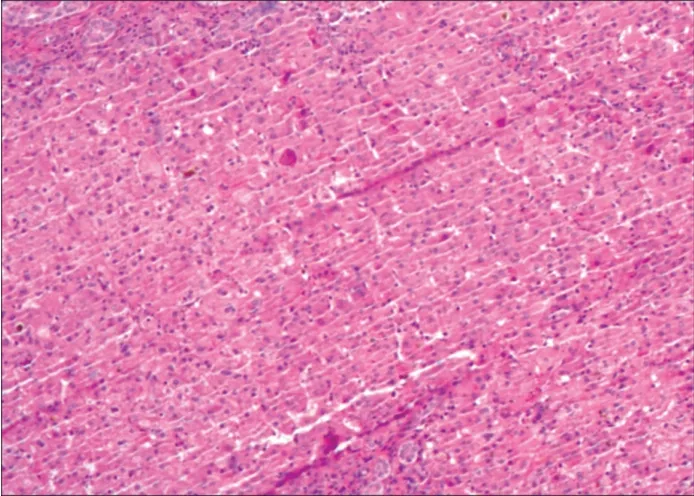
图3 本例患儿术中肝活检病理图示肝纤维化3 级(HE染色,×200)
BASM常因合并其他系统畸形,肝门-空肠吻合手术难度较大,疗效亦较差,且后期肝移植难度亦较大。BASM患儿出生后即发生难治性黄疸,且呈进行性加重,早期明确诊断且及早行肝门-空肠吻合手术可避免不可逆的胆汁性肝硬化。本例患儿于出生后54 d 行肝门-空肠吻合术,术后胆红素下降明显,肝功能趋于正常,术后1 年随访患儿体格发育良好,神经精神测评正常。
总之,超声在诊断BASM 及术后随访中均有一定的临床价值,为临床早期干预争取了时间。
猜你喜欢
心电与循环(2021年4期)2021-11-29
云南医药(2021年3期)2021-07-21
散文诗世界(2019年6期)2019-09-10
中国临床医学影像杂志(2019年6期)2019-08-27
意林·全彩Color(2019年7期)2019-08-13
中学数学杂志(2019年9期)2019-05-29
中国眼镜科技杂志(2016年17期)2016-10-24
西南医科大学学报(2015年1期)2015-08-22
肝胆胰外科杂志(2015年5期)2015-02-27
西南军医(2014年5期)2014-04-25
