
类石墨烯氮化碳制备及其在气体分离领域中应用的研究进展
2023-12-04 08:54:56徐双平贾宏葛张明宇蘧延庆徐靖宇
中国塑料 2023年11期
张 超,徐双平*,贾宏葛,张明宇,蘧延庆,徐靖宇
(1.齐齐哈尔大学材料科学与工程学院,黑龙江省聚合物基复合材料重点实验室,黑龙江 齐齐哈尔 161006;2.大连工业大学轻工与化学工程学院,造纸工程重点实验室,辽宁 大连 116034)
0 前言
随着现代工业的发展,全球的能源消耗在日益增加,最为突出的是化石燃料的燃烧造成了CO2等气体的排放量急剧增长,造成臭氧层的破坏,对现代社会的可持续发展构成了巨大的威胁。传统的气体分离技术有着能耗高、成本高等缺点,气体膜分离与其相比具有节能、高效、无二次污染等优点[1]。g-C3N4纳米材料制备气体分离膜是一个简单、经济、绿色环保的过程,与碳纳米管和石墨烯制备过程相比,g-C3N4的制备简单,是制备聚合物混合基质气体分离膜的理想填料。
C3N4是一种无机非金属材料,被认为是年代久远的人工化合物之一,其历史可以追溯到19世纪初期。1830年,Berzelius就报道了这种物质,后来,Liebig在聚合氯化铝和硫氰酸钾时发现了黄色固体粉末,他将其命名为“melon”[2],Liebig 发现的melamine、melem、melam 和melon 被确认为基本结构单元是三-s-三嗪和三嗪。但是很不幸,melon这种材料并没有推动自身的发展。直到1922年,Franklin[3]分析了这种材料,他表明,经过聚合一系列的氨碳酸,可以形成C3N4。后来在1937年,Pauling 和Sturdivant[4]通过X 射线晶体学再次确认了三-s-三嗪是melon 的结构单元。C3N4不能溶于其他的化学试剂,从而导致其研究进展极其缓慢。1989年,Cohen和Liu[5]得到了β-C3N4,预测其硬度与金刚石相近,从此科研人员对C3N4的研究逐渐变得狂热。在1996年,Teter 和Hemley[6]发现了5 种类型的C3N4,分别为α-C3N4、β-C3N4、假立方C3N4、立方C3N4和g-C3N4。碳化氮是碳和氮的化合物,而g-C3N4是碳氮化合物中最稳定的同素异形体之一。目前为止,g-C3N4已被用于不同工艺的膜改性,气体分离膜是其中的一种。g-C3N4因其有机结构,可以很好地分散在聚合物基体当中,比无机纳米薄片作为填料例如沸石纳米颗粒更加合适。
近些年来,膜分离技术的发展在气体分离领域取得了显著的进步,膜分离技术具有占地面积小、比表面积大、环境友好、成本低、节能等特点,在环境保护和修复中具有广阔的应用前景,g-C3N4及其功能化纳米复合材料被认为是下一代环境友好分离膜的有吸引力的候选材料[7]。g-C3N4加入到基质膜中在增强膜分离性能的同时,也存在着其他问题使混合基质膜(MMMs)达不到理想的性能,包括g-C3N4和聚合物之间的相容性以及g-C3N4作为填料在MMMs 的制备过程中发生团聚堵塞孔隙导致气体分离性能变差。因此,需要选择合适聚合物与g-C3N4共混来开发具有高气体分离性能和高力学强度的MMMs[8]。
1 氮化碳
1.1 g-C3N4的结构和性质
1.1.1 结构
经研究表明,g-C3N4是一种近似石墨烯的平面二维片层结构,其结构有2 种基本单元[9],图1 显示了主要的构造单元三嗪(C3N3)和三-s-三嗪环(C6N7)结构,形成g-C3N4的异构体。三嗪和三-s-三嗪环分别以基本结构单元无限延伸形成了网状结构,二维纳米片层间通过范德华力结合[10]。Kroke 等[11]经过密度泛函理论(DFT)计算得出三-s-三嗪环结构单元比三嗪环结构单元连接而成的g-C3N4要更加稳定。Ma 等[12]研究了纯六方庚嗪基g-C3N4及其掺杂体系,最稳定的石墨相将以第36 号空间群cmc21 的六方结构结晶,它由氮连接的庚烷单位组成,以类似石墨的结构以ABAB 的方式交错排列,如图2所示。所有的C 原子都经历了3 个N原子的三重配位,桥上的N 原子和内部的N 原子都被3 个C 原子三重配位,边缘的N 原子被2 个C 原子二重配位。

图1 g-C3N4的结构Fig.1 Structures of g-C3N4

图2 g-C3N4的层间结构[12]Fig.2 Interlayer structure of g-C3N4[12]
1.1.2 性质
g-C3N4的物理性质非常稳定,因骨架上含有芳香杂环结构从而具有很好的热稳定性,经过测试g-C3N4在600 ℃单位高温条件下也能保持原有结构而不发生分解。具有层状结构通过分子间作用力连接起来的二维材料具有非常稳定的化学性质,g-C3N4是二维层状材料的经典代表之一。有研究人员测试了g-C3N4在各种有机溶剂中的稳定性和耐久性,Wang 等[13]将g-C3N4粉末分散在水、丙酮、乙醇、吡啶、乙腈、二氯甲烷、N,N-二甲基甲酰胺(DMF)、冰醋酸、0.1 M NaOH 水溶液中30 d,测定这些被浸泡在各种有机溶剂中g-C3N4粉末的红外光谱,与未用各种试剂浸泡过的g-C3N4粉末进行红外光谱比较,二者红外光谱基本相同,证明没有发生化学变化。
1.2 g-C3N4的制备方法
g-C3N4的制备方法有很多,主要包括热聚合法、溶剂热法、固相反应法、化学气相沉积法等。制备g-C3N4的前驱体种类多,如尿素、硫脲、三聚氰胺等,且价格低廉,适合大规模生产。每种制备方法都有其优点和缺点。不同的制备方法可以形成不同形貌的g-C3N4,具有不同的理化性质。
1.2.1 热聚合法
热聚合法是通过用加热的方法将单体激发成为单体自由基,从而引发单体聚合的一种自由基型聚合方法,直接加热尿素、三聚氰胺等含有氰胺结构的有机物来制备g-C3N4。以尿素前驱体制备g-C3N4为例,随着反应温度的升高,热聚合过程中形成的尿素残留物从尿素变为双缩脲、三聚氰胺、氨酰、氨酰和三聚氰胺(图3),在加热过程开始约152 ℃时,尿素分解的产物充满了整个容器,产物包括氰酸和氨,在大约160 ℃下氰酸与完整的尿素反应生成双缩脲,然后双缩脲与氰酸反应生成三聚氰酸,三聚氰酸再与NH3反应生成氨酰,随着反应温度的进一步升高,由氨酰胺与NH3反应得到氨酰胺,再由氨酰胺与NH3进一步反应生成三聚氰胺,最后通过三聚氰胺固相共聚,在小坩埚中合成了块体g-C3N4;氰酸可以在175 ℃左右的气相条件下自发快速聚合生成氰尿酸,氰尿酸与NH3反应生成氨酰,氨酰与NH3进一步反应得到三聚氰胺,在三聚氰胺的气相共聚过程中,形成的大量NH3嵌入到三聚氰胺的分子空间中,破坏了脆弱的范德华键,随着温度的降低,气态产物落入坩埚中,最后在较大的陶瓷坩埚中获得超薄g-C3N4纳米片[14]。
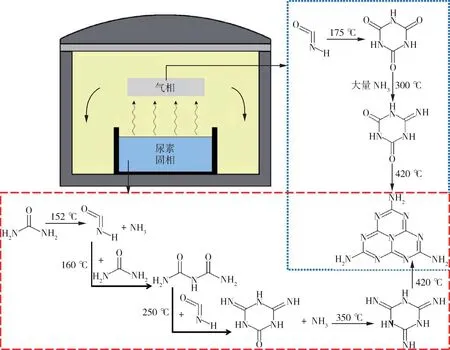
图3 g-C3N4在气相和固相的不同热聚合过程[14]Fig.3 Different thermal polymerization processes of g-C3N4 in gas and solid phase[14]
Liu 等[15]在标准大气和没有添加剂辅助情况下通过尿素的热解制备的石墨相氮化碳,将尿素干燥24 h后放入烘箱加热至550 ℃恒温3 h,最终得到了黄色的g-C3N4。他们还研究了温度对以尿素为前驱体制备的g-C3N4结构的影响,结果表明,类似石墨的层状结构首先形成,然后,在高于450 ℃的温度下发生了平面内排序的构建。Sun 等[16]将尿素颗粒在110 ℃下溶解在乙醇中进行预处理,以20 ℃/min 的加热速率加热至550 ℃恒温2 h 后得淡黄色的碳化碳纳米片,制备过程如图4所示。Yan 等[17]在半封闭体系中直接加热三聚氰胺,在马弗炉中以20 ℃/min 的加热速度加热到,500 ℃并在500、520、550 和580 ℃下分别进行2 h 的进一步脱氨处理,其DSC 曲线显示,制备的g-C3N4在600 ℃开始分解,分解产物立即燃烧。在加热g-C3N4的过程中没有观察到脱氨过程,三聚氰胺与g-C3N4的DSC 曲线相比,可以确认三聚氰胺加热过程中630 ℃的内热峰应归因于三聚氰胺热解过程中形成的g-C3N4的分解,这比制备的g-C3N4的起始分解温度(600 ℃)高30 ℃,这表明,g-C3N4在半封闭的氨气环境中比在开放系统中更稳定。而Zhai等[18]在实验中加热三聚氰胺的温度范围为500~650 ℃,加热速率为5 ℃/min,随着热解温度的升高,粉末产物的颜色变深,根据已有报道,当温度升高时,热解所得到产品的结晶会更完美。Zhou 等[19]开发了一种无模板的水热辅助热聚合方法,用于大规模合成一维石墨氮化碳微管。g-C3N4微管是由三聚氰胺-氰尿酸复合微棒在N2氛围下进行简单的热聚合而得到的,该微米棒是由三聚氰胺溶液在180 ℃下水热处理24 h 而合成的,得到的g-C3N4微管具有较大的表面积和独特的一维管状结构,扫描电子显微镜(SEM)照片如图5所示。Zhang 等[20]将三聚氰胺与硫脲以2/1 的质量比溶于一定量乙醇溶剂中,80 ℃条件下连续搅拌1 h,通过重结晶将乙醇蒸发后得到尿素与硫脲的混合物,以5 ℃/min的加热速率升温至400 ℃,恒温5 h后再以5 ℃/min 的加热速率升温至600 ℃持续4 h 得到了g-C3N4粉末。Qin 等[21]将制备的三聚氰胺和硫脲复合材料作为前驱体,在5 ℃/min 下加热至600 ℃恒温4 h 后得到了具有高比表面积的薄层g-C3N4纳米片,经研究发现,通过改变硫脲在复合前驱体中的摩尔百分比,可以调节所制备的g-C3N4的结构、形态、光学性能和光催化活性。

图4 热聚合法g-C3N4纳米片的制备过程[16]Fig.4 Preparation process of g-C3N4 nanosheets by thermal polymerization[16]

图5 复合微棒和一维g-C3N4微管的SEM照片[19]Fig.5 SEM images of complex microrods and 1D g-C3N4 microtubes[19]
1.2.2 溶剂热法
溶剂热法在早期主要用于合成非类石墨相氮化碳,是一种在密闭体系中以有机物作为溶剂,在一定温度和压力下,使原始物质进行反应的一种合成方法。溶剂热法制备g-C3N4的过程简单且易于控制,具有反应条件温和、体系均匀性好、隔绝外界环境因素等优点[22]。近年来,人们将溶剂热法的首选目标确定为g-C3N4,为的是探索不同前驱体以及不同有机溶剂制备g-C3N4的可行性。例如Montigaud 等[23]以三聚氰胺为原材料,以肼(NH2NH2)为溶剂,在高温高压(800~850 ℃,3 GPa)条件下合成了g-C3N4,并通过X 射线衍射仪(XRD),SEM,电子探针微分析仪,化学分析电子能谱(ESCA),红外光谱仪(FTIR)等对g-C3N4进行了表征。紧接着Montigaud 团队[24]将三聚氰胺和氰尿酰氯在超临界状态(130 MPa,250 ℃)下以三乙胺(Et3N)作为溶剂进行缩合得到了g-C3N4固体,三聚氰胺和氰尿酰氯的结构可以很容易地通过缩合诱导出预期的腔隙石墨形态,缩合过程如图6所示。以三聚氰胺和氰尿酰氯缩合获得的固体虽然呈现石墨型结构,但是结晶度较低,稳定性较差。Bai等[25]以常见的化合物四氯化碳和氯化铵为原料,在高压釜中加热至400 ℃持续24 h,且在无催化剂条件下合成了类石墨C3N4纳米晶体,所得g-C3N4结晶良好,还获得了一些不同尺寸的单晶。Luv 等[26]将1,3,5-三氯三嗪(C3N3Cl3)和氮化锂(Li3N)分别作为碳源和氮源,以苯作为反应溶剂,在高压反应釜中以360 ℃和6~7 MPa 条件下恒温12 h,经过处理最终得到了深棕色产物,并用XRD、X 射线光电子能谱仪(XPS)和FTIR 对产物进行了表征。Andreyev 等[27]选择的化合物较为复杂且不常见,他们将六氯苯(C6Cl6)作为溶剂和反应物,与Na3N 在高温高压(400~500 ℃,7.7 GPa)条件下保持50~70 h,得到了黑褐色的产品。
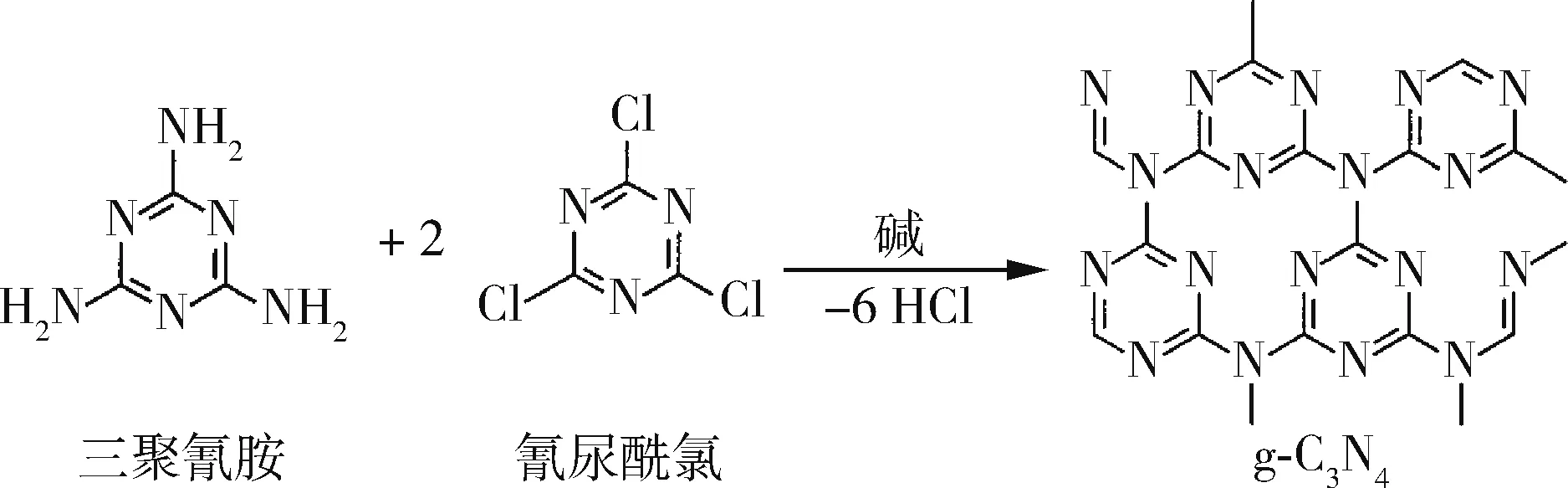
图6 三聚氰胺与氰尿酰氯的缩合[24]Fig.6 Condensation of melamine with cyanuryl chloride[24]
溶剂热法在能提供良好反应环境的同时,还能通过改变反应条件控制反应物分子的自组装过程,能得到具有特殊形貌结构的g-C3N4材料。例如Li等[28]以NiCl2为催化剂前驱体,在230 ℃和1.8 MPa条件下,三聚氯氰与Na 反应形成氮化碳纳米管。Bai 等[29]则以三聚氰胺作为前驱体,四氯化碳作为溶剂,向混合体系中加入粒径80~100 nm 的SiO2球作为模板剂,高压釜在250 °C 和4~5 MPa 条件下保持24 h,利用稀氢氟酸去除SiO2模板剂干燥后得到了黑色粉末。
1.2.3 固相反应法
固相反应是固体间发生化学反应并生成新固体产物的过程,常用来制备g-C3N4。制备氮化碳时常选用含有三嗪结构的化合物,例如三聚氰胺、三聚氰氯等。Guo 等[30]选择三聚氰氯作为前驱体,与NaNH2、K 和NaN3分别在高压釜中发生反应,3 个反应釜分别在220 ℃下恒温8 h,300 ℃下恒温20 h,380 ℃下恒温12 h,自然冷却到室温后得到棕黑色固态产品。Gu等[31]也是选择三聚氰氯作为前驱体,与氰氨化钙发生固相反应,在高压反应釜中以500~650 ℃的反应温度保持10 h,能得到结晶的g-C3N4,但遗憾的是产品纯度较低,其中含有大量的石墨碳。Khabashesku 等[32]以三聚氰氯为前驱体,与氮化锂发生固相反应,加热至380 ℃并保持2~4 h,反应结束除去副产物氯化锂得到了深棕色产物。Zhang 等[33]所采用的前驱体和氮源都是含有三嗪结构的化合物三聚氰胺和三聚氰氯,将三聚氰胺与三聚氰氯以1/1 的摩尔比进行混合在1.0~1.5 GPa、500~550 ℃条件下固相反应制备了化学计量结构为C6N9H4Cl 的新型氮化碳材料。Li 等[34]也是选用三聚氰胺和三聚氰氯这两种化合物作为反应物,在负压状态下,加热至150 ℃约24 h,然后在30 min 内将反应室加热至800 ℃保持2 h后得到具有空心结构的g-C3N4纳米棒,其透射电子显微镜(TEM)照片如图7所示。Tragl 等[35]发现利用固相反应能控制反应产物的结构,可以得到特殊形貌的g-C3N4纳米片,他们通过三聚氰氯与不同氮源[如Li2(CN2)或Li3(BN2)]的复分解反应合成了非晶态氮化碳材料的微管和纳米管。Zimmerman 等[36]通过模板导向的三聚氰氯或三聚氰胺与氮化锂的固相反应制备了直径从20 μm 到30 nm 不等的氮化碳球。Lu 等[37]首次在Zn 粉作为催化剂下,在40 MPa 和220 ℃下合成了氮化碳纳米线和假立方体C3N4多晶纳米颗粒。

图7 样品的TEM照片[36]Fig.7 TEM images of the samples [36]
1.2.4 化学气相沉积法
Tanaka 等[38]用微波等离子体化学气相沉积(CVD)法对氮化碳的低温合成进行了研究,以硅作为衬底,CH4和N2作为反应气体,压力在1.1~4.0 kPa 之间变化,微波功率在400~800 W 之间变化,用改进的微波等离子体CVD 装置合成了氮化碳,他们经过研究发现沉积物的形态依赖于衬底温度,并且在低至467 ℃的衬底温度下获得面状颗粒。Kosaka等[39]使用蓝宝石作为衬底,以三聚氰胺为前驱体,将粉末加热到500~650 ℃,在由管式炉组成的定制CVD 设备中制备了g-C3N4薄膜。Yadav 等[40]选择双氰胺(C2H4N4)为前驱体作为C 源和N 源,放进石英管入口处,即通入氩气的一端,末端与真空泵相连,如图8中所示,由于氩气流和石英管内的低真空,双氰胺粉末的蒸汽进入600 ℃熔炉,在高温下的冷凝产生了沉积在石英管内的g-C3N4结构。Chamorro-Posada 等[41]将三聚氰胺氰尿酸盐粉末置于陶瓷坩埚底部,顶部放置1 个25 m2正方形陶瓷玻璃板后封闭坩埚,在马弗炉中加热至600 ℃持续30 min后,在陶瓷玻璃面上得到g-C3N4膜。Chubenko 等[42]将双温区开启式管式炉作为实验仪器,将三聚氰胺粉末装入“低温”区石英管内,将玻璃和硅衬底水平放置在预期的“高温”区,以150 cm3/min 的气流向炉管中充入干燥的氩气,将“高温”区加热至500~650 ℃范围内,低温区的温度会被带动到350 ℃左右,待体系稳定后得到g-C3N4膜,在玻璃和硅衬底上合成的所有薄膜都是连续均匀的,表面相当光滑,并研究了550、600、650 ℃下不同衬底温度对薄膜形态的影响,发现薄膜呈现出轻微的波状形态,在图9 的SEM 照片中可以看出合成温度为600 ℃时薄膜厚度最大,玻璃薄膜厚度为1.15 μm,硅薄膜厚度为0.90 μm。

图8 以双氰胺为前驱体CVD法制备g-C3N4[40]Fig.8 g-C3N4 prepared by CVD method using dicyanamide as precursor[40]

图9 不同温度下玻璃和硅衬底上合成的不同厚度薄膜的SEM照片[42]Fig.9 SEM images of films of different thickness synthesized on glass and silicon substrates at different temperature[42]
针对上述4 种g-C3N4的合成方法对合成产物的影响以及各自优缺点汇总如表1所示。

表1 不同制备方法对g-C3N4性能的影响Tab.1 Effects of different preparation methods on properties of g-C3N4
2 基于g-C3N4气体分离膜
g-C3N4纳米材料已经在气体分离领域中得到了广泛的应用,聚合物/g-C3N4混合基质膜(MMMs)常见的混合基质膜有自具微孔聚合物(PIM)/g-C3N4膜、聚醚嵌段聚酰胺(Pabax)/g-C3N4膜、g-C3N4离子液体支撑液膜(SILM)等。
2.1 PIM-1/g-C3N4 MMMs
制备性能优良的混合基质膜,选择合适的聚合物材料作为基质是非常重要的。PIM 具有非常好的气体渗透性,是一种相对比较好的聚合物材料,因此非常适合用来做混合基质膜的基质。Tian 等[43]选择了研究最多的且容易合成的PIM-1,首先将PIM-1溶解在氯仿中形成原液,g-C3N4也分散在氯仿中并超声12 h,然后将一定量的g-C3N4氯仿分散体加入PIM-1储备溶液中并搅拌过夜,再将浇铸溶液倒入聚四氟乙烯盘中,并在环境温度下的氮气中蒸发得到了MMMs。g-C3N4在PIM-1/g-C3N4MMMs 中表现出明显的气体输送作用,与纯PIM-1膜相比,在相对较低的g-C3N4负载下,MMMs的透气性大大提高,具有粒度筛分作用的g-C3N4更有利于小分子(特别是H2)的传输,H2/CH4和H2/N2的选择性增加,且气透性不受影响,同时g-C3N4的加入也改善了MMMs 的热性能和力学性能。Voon 等[44]通过4 种不同的合成方法对g-C3N4纳米片进行了硫酸基、脂肪族氨基、芳香族氨基和磺酸基等官能团的改性,如图10所示,将改性后的4种g-C3N4纳米片分别与PIM-1共混以制备共混基质膜。他们总结出与胺改性的g-C3N4相比,具有磺化g-C3N4的MMMs 显著增强了气体传输性能,特别是对于CO2分离,这表明磺酸基团对CO2的亲和力更强,并诱导了与CO2分子更大的偶极-偶极相互作用,5 %(质量分数,下同)g-C3N4-D 的MMMs 超过了2008年O2/N2和H2/N2分离的Robeson界限(图11),表明PIM-1 和磺化g-C3N4MMMs 在CO2捕获、H2分离和空气分离方面具有潜力,另外发现g-C3N4-D 嵌入PIM-1基质对抑制物理衰老也有积极作用。

图10 g-C3N4纳米片的胺修饰(g-C3N4-A/B/C)和磺化(g-C3N4-D)过程[44]Fig.10 Amine modification(g-C3N4- A/B/C)and sulfonation(g-C3N4-D)process of g-C3N4 nanosheets[44]

图11 膜O2/N2和H2/N2分离的Robeson界限[44]Fig.11 Robeson bounds of O2/N2 and H2/N2 separation of the samples[44]
2.2 Pebax/g-C3N4 MMMs
Pabax 是一种介于热塑性弹性体和橡胶体之间的树脂,性能优异且制造简单。Cheng 等[45]将一定的g-C3N4纳米片通过轻度超声溶解在乙醇和水(体积比7/3)的混合溶剂中,然后加入Pebax,在80 ℃下用冷凝器管搅拌12 h,将铸造液以1 000 r/min 的速度在PAN 基板上进行旋转镀膜,制备了不同负载以及掺入不同形态g-C3N4纳米片的MMMs。g-C3N4的多孔结构为CO2分子提供了良好的运输通道(图12),使Pebax/g-C3N4MMMs 具有良好的CO2/N2分离性能,CO2渗透系数为33.3 GPU,CO2/N2选择性高达67.2,比纯的Pebax 膜提高了100 %,g-C3N4纳米片在CO2吸附和分子筛分性能上的差异使膜具有区域吸附系数和扩散系数,从而显著提高了分离性能。而Guo等[46]以具有筛分孔结构的g-C3N4纳米片为模板,与沸石咪唑酯骨架(ZIF-90)相结合构建了零维/二维复合材料ZIF-90@C3N4(ZCN),他们将Pebax-1657 溶于水与乙醇体积比3/7的混合溶剂中,将Pebax 溶液与分散在混合溶剂中的ZCN 填料混合来制备用于MMMs的铸膜液,将铸膜液涂抹在聚四氟乙烯板上后得到了Pebax/ZCN MMMs。显著改善了膜内纳米片的堆积和收缩现象,当ZCN 负载量为8.0 %时,分离性能最佳,CO2渗透率高达110.51 Barrer,CO2/N2选择性为84.35,分别比原始Pebax 膜提高了75.7 %和41.6 %,气体分离性能远高于g-C3N4/Pebax MMMs,其CO2/N2气体分离性能如图13所示,超过了2008年Robeson上限。
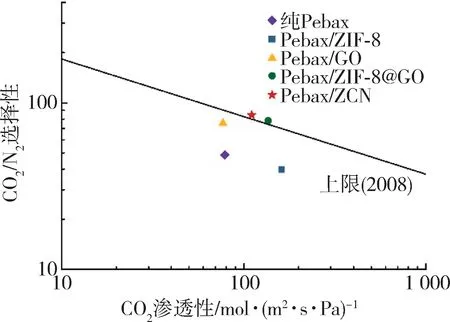
图13 Pebax/ZCN MMMs在2008年Robeson上限图中的位置[46]Fig.13 Position of Pebax/ZCN MMMs in the 2008 Robeson upper limit diagram[46]
2.3 g-C3N4 SILM
支撑液膜(SLM)具有高CO2渗透速度和分离因子,通过将亲CO2溶剂嵌入多孔基质中而制成,并广泛用于分离CO2[47]。SLM 由有机阳离子和有机或无机阴离子组成,具有高二氧化碳容量,热稳定性好,被广泛用于制备用于气体分离的SILMs[48]。Niu 等[49]通过将纳米离子液体嵌入2D g-C3N4纳米通道制备的气体分离膜,用于高效分离CO2/N2和CO2/CH4,其制备和气体渗透过程如图14所示。通过使用磁力搅拌将一定量的g-C3N4纳米片分散在乙醇和水的混合溶剂中,使用真空过滤装置在聚四氟乙烯(PTFE)基底膜上真空过滤该均匀溶液以获得g-C3N4层压膜,在40 ℃的真空烘箱中干燥8 h 后将g-C3N4层压膜上以1 000 r/min 多次旋涂不同浓度的1-乙基-3-甲基咪唑乙酸酯([EMIm][AcO],IL),旋涂过程中,离子液体通过强大的毛细管力逐渐进入g-C3N4的2D 纳米通道,最终被均匀地限制在纳米通道中形成g-C3N4薄膜。由于离子液体的引入,通过溶解-扩散过程,制备的无缺陷g-C3N4SILMs与未掺加IL 的g-C3N4薄膜相比,气体选择性和渗透性显著提高,g-C3N4SILM 除了具有优异的选择性外,还表现出特殊的稳定性和耐久性,在图15 中,g-C3N4SILM 与已报道的二维材料基膜、IL 基膜、SILM 及其他膜的气体分离性能进行了比较,制备的g-C3N4SILMs 具有良好的CO2渗透性,并能保持良好的CO2/N2和CO2/CH4选择性,他们表明在二维g-C3N4层压膜中进行纳米约束是提高气体分离性能的有效途径。

图14 g-C3N4 SILM制备及气体渗透过程示意图[49]Fig.14 Schematic diagram of g-C3N4 SILM preparation and its gas penetration process[49]

图15 g-C3N4 SILM与其他种类膜气体分离性能的比较[49]Fig.15 Comparison of gas separation properties of g-C3N4 SILM with other species of membrane[49]
2.4 聚酰亚胺(PI)/g-C3N4 MMMs
PI有着良好的成膜性,PI膜的力学性能好、热稳定性高,分离性尤为突出。Soto-Herranz等[50]将化学改性的g-C3N4作为表面活性剂添加到商业聚酰亚胺(Matrimid)膜中进行修饰,制备出了0.5 %和2 % g-C3N4负载的Matrimid/g-C3N4MMMs,他们对g-C3N4进行了质子化、羟基化和肼化(图16),其中,2 %羟基化g-C3N4的MMMs表现出最佳的CO2/CH4分离性能,与纯Matrimid 膜相比提高了52.2 %,0.5 %质子化g-C3N4的MMMs 对CO2/CH4的选择性与纯Matrimid 膜相比提高了36.9 %。改性的g-C3N4与Matrimid 基体之间有着较强的界面相互作用,使MMMs 具有很高的抗溶胀性和力学稳定性。他们将在Matrimid 膜中掺入改性的g-C3N4可以提高气体分离性能归因于Matrimid/g-C3N4中氢键的增加或者由g-C3N4介导的Matrimid中的交联修饰。

图16 g-C3N4的化学改性[50]Fig.16 Chemical modification of g-C3N4[50]
2.5 聚醚砜(PES)/g-C3N4 MMMs
聚醚砜(PES)膜有着很好的稳定性,可在碱性和酸性环境中保持稳定,耐热性好,在200 ℃以内机械性能基本没有变化。Jomekian 等[51]用壳聚糖(CS)修饰的g-C3N4分散在流延的PES 溶液上,作为ZIF-8 颗粒生长的平台,利用相转化法将PES 铸膜液浸入水浴中24 h。采用逐层结晶法在PES 载体上合成了PES /g-C3N4/ZIF-8 MMMs,将六水合硝酸锌和改性的g-C3N4分散在甲醇和水的混合体系中,搅拌后转移到装有PES 膜的容器中陈化5 min,再用2-甲基咪唑甲醇和水的混合溶液替代上述混合液陈化5 min,将这2 个过程按顺序循环4 次作为1 个周期,9 个周期后将MMMs取出干燥后得到了PES/g-C3N4/ZIF-8 MMMs。CS改性的PES/g-C3N4/ZIF-8 MMMs 的CO2/CH4选择性是PES/ZIF-8 MMMs 的10.5 倍,随着改性g-C3N4的加入提高了膜的CO2/CH4选择性,主要因为CS中含有大量氨基对CO2有很强的亲和力。此外,作者利用红外光谱和核磁共振分析证明了g-C3N4和ZIF-8 的咪唑基团之间形成了C-N 键,使改性后的PES/g-C3N4/ZIF-8 MMMs柔韧性显著提高。
针对上述多种掺有类石墨烯氮化碳的混合基质膜各自的应用特点进行了介绍并且在表2中汇总。

表2 不同MMMs的应用特点Tab.2 Application characteristics of different MMMs
3 结语
g-C3N4作为一种类石墨烯的二维结构,同时其优异的柔韧性,利于制备具有单原子层的纳米片,使其具有高比表面积。基于g-C3N4其柔韧性和高比表面积在堆砌时有利于片与片之间紧密平铺,这将有助于制备超薄膜,气体传输时能有效减少传质距离,从而提高膜对气体的渗透性能。然而,相较于其他二维材料在气体膜分离领域的应用,g-C3N4不仅是在数量上,还是性能上都有所欠缺。因此,可以借鉴现有二维材料在高性能气体分离膜材料制备方法,以及遵循可持续发展的原则,从以下几方面进行改性制备g-C3N4基气体分离膜:
(1)基于薄层复合膜的模块化构筑理念,以商品化PI、聚丙烯腈、聚砜、PES、PEI 和聚偏氟乙烯等多孔材料为底层支撑层;g-C3N4片为分离层,采用诸如溶液涂敷、粉末喷涂,以及沉积等方法进行附层;具有高气体透过性的聚硅氧烷为中间层能有效防止g-C3N4片入侵基底多孔层,同时起到底层和分离层间的黏结层;
(2)混合基质膜作为分离膜的一大类,因具有生产工艺简便、周期短、成本低等优点,被广泛应用;可以根据分离环境、分离对象,有所侧重选择合适的聚合物为基体,g-C3N4片为分散相,制备混合基质,不失为未来工业膜研发的方向;
(3)遵循可持续发展理念,对生物质材料功能化改性以满足更高性能的需求;诸如,纤维素及其衍生物作为最早的膜材料之一,由于其自身结构的限制,导致其耐温和耐化学性不高,同时气体分离性能也不佳;因此,可以对其进行结构改性,在结构中引入氨基、更多羟基和羧基,然后和有机化修饰后的g-C3N4片通过接枝反应制备g-C3N4接枝纤维素膜,将有望改善其性能,满足当下气体膜分离领域的需求。
猜你喜欢
中国化肥信息(2022年2期)2023-01-02 12:17:29
中国化肥信息(2022年8期)2022-11-30 06:20:14
陶瓷学报(2021年1期)2021-04-13 01:33:08
热处理技术与装备(2019年1期)2019-03-14 08:07:20
中国化肥信息(2018年2期)2018-08-23 09:09:16
电子制作(2018年12期)2018-08-01 00:47:48
咸阳师范学院学报(2016年6期)2017-01-15 14:18:45
上海金属(2016年2期)2016-11-23 05:34:32
中国化肥信息(2016年41期)2016-05-17 04:25:58
合成化学(2015年1期)2016-01-17 09:01:13
