
实脾消水凝胶贴膏的薄层色谱鉴别和含量测定研究
2023-12-02 10:26马秉智梁莹莹王海洋唐永和中日友好医院药学部北京100029
药学实践杂志 2023年11期
马秉智,梁莹莹,王海洋,唐永和,李 栋,赫 军 (中日友好医院药学部, 北京 100029)
实脾消水浸膏为中日友好医院的自制中药制剂,来源于中西医结合肿瘤内科李佩文教授的经验方“消水Ⅱ号”,处方由黄芪、车前子、红花等组成,具有益气活血、渗湿利水的功能,临床用于癌性胸腹水的辅助治疗,能明显的延长晚期肿瘤患者的生存期,提高晚期肿瘤患者的生存质量[1-3]。到目前为止,该制剂在院内已使用近三十年,由于这种浸膏剂使用不方便,因此,根据临床需要,本课题组将其改为凝胶贴膏剂。为了更好地控制它的质量,本文采用薄层色谱法(TLC)对其中的黄芪、车前子、莪术、桂枝、猪苓、预知子进行定性鉴别,采用超高效液相色谱-串联质谱法对制剂中君药黄芪所含成分黄芪甲苷进行含量测定,现报道如下。
1 仪器与试药
1.1 仪器
Ultimate 3 000 高效液相色谱仪、Q ExactiveTM组合型四级杆Orbitrap 质谱仪(赛默飞公司);BSA224S 型电子天平(德国赛多利斯仪器系统有限公司);DZKW-4 电子恒温水浴锅(北京中兴伟业仪器有限公司);RE-52AA 旋转蒸发器(上海亚荣生化仪器厂);ZF-90 型暗箱式紫外投射仪(上海顾村电光仪器厂);硅胶 G 薄层板(青岛海洋化工分厂)。
1.2 试药
实脾消水凝胶贴膏(自制,批号:20180102、20180115、20180203);黄芪甲苷(批号:110781-201616)、京尼平苷酸(批号:111828-201604)、毛蕊花糖苷(批号:111530-201713)、吉马酮(批号:111665-201605)、麦角甾醇(批号:111845-201403)、桂枝对照药材(批号:121191-201605)、预知子对照药材(批号:121492-201102)购自中国食品药品检定研究院;乙腈为色谱纯,水为高纯水,其他试剂均为分析纯。
2 方法与结果
2.1 TLC 鉴别
2.1.1 黄芪
取本品1 片,除去保护膜后,称取药膏10 g,加甲醇100 ml,加热回流1 h,滤过,滤液蒸干,残渣加水30 ml 使溶解,用水饱和正丁醇振摇提取2 次,每次30 ml,合并正丁醇液,用水洗涤2 次,每次20 ml,弃去水液,正丁醇液蒸干,残渣加甲醇5 ml 使溶解,加于中性氧化铝柱(100~120 目,5 g,内径为10~15 mm)上,用40%甲醇100 ml 洗脱,收集洗脱液,蒸干,残渣加甲醇0.5 ml 使溶解,作为供试品溶液。另取黄芪甲苷对照品,加甲醇制成每1 ml含1 mg 的溶液,作为对照品溶液。取除黄芪外的其他药材制成凝胶贴膏,按照上述方法制备黄芪的阴性对照溶液。按照《中国药典》2020 年版的薄层色谱法[4],吸取供试品溶液、阴性溶液各6 μl,黄芪甲苷对照品溶液1 μl,分别点于同一硅胶G 薄层板上,以三氯甲烷-甲醇-水(13:5:0.6)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的橙黄色荧光斑点,且阴性无干扰,结果见图1A。
2.1.2 车前子
取本品1 片,除去保护膜后,称取10 g,加甲醇100 ml,加热回流1 h,滤过,滤液蒸干,残渣加热水20 ml 使溶解,加于D101 型大孔吸附树脂柱(内径为1.5 cm,柱高为12 cm)上,用水100 ml 洗脱,弃去水液,再用30%乙醇100 ml 洗脱,收集洗脱液,蒸干,残渣加甲醇2 ml 使溶解,作为供试品溶液。另取京尼平苷酸、毛蕊花糖苷对照品,加甲醇分别制成每1 ml 含1 mg 的溶液,作为对照品溶液。再取除车前子外的其他药材制成凝胶贴膏,按照上述方法制备车前子阴性溶液。按照《中国药典》2020 年版的薄层色谱法,吸取上述三种溶液各10 μl,分别点于同一硅胶 GF254薄层板上,以乙酸乙酯-甲醇-甲酸-水(18:2:1.5:1)为展开剂,展开,取出,晾干,喷以2%香草醛硫酸溶液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,且阴性无干扰,结果见图1B。
2.1.3 莪术
取本品1 片,除去保护膜,称取药膏10 g,加甲醇100 ml,加热回流1 h,过滤,滤液挥干,残渣加水30 ml 使溶解,加石油醚(30~60 ℃)振摇提取2 次,每次20 ml,合并石油醚液,蒸干,残渣加无水乙醇4 ml 使溶解,作为供试品溶液。另取吉马酮对照品,加无水乙醇制成每1 ml 含0.4 mg 的溶液,作为对照品溶液。另取除莪术外的其他药材制成凝胶贴膏,按照上述方法制备莪术阴性对照溶液。按照《中国药典》2020 年版的薄层色谱法,吸取上述三种溶液各10 μl,以石油醚(30~60 °C)-丙酮-乙酸乙酯(94∶5∶1)为展开剂,点于同一硅胶G 薄层板上,展开,取出,晾干,喷以2 %香草醛硫酸溶液,在105 °C 加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,且阴性无干扰,结果见图1C。
2.1.4 桂枝
取本品1 片,除去保护膜,称取药膏10 g,加甲醇100 ml,加热回流1 h,滤过,滤液挥干,残渣加热水30 ml 使溶解,加三氯甲烷振摇提取2 次,每次30 ml,合并三氯甲烷液,蒸干,残渣加乙酸乙酯2 ml使溶解,作为供试品溶液。另取桂枝对照药材2 g,加乙醚10 ml,浸泡30 min,时时振摇,滤过,滤液挥干,残渣加三氯甲烷l ml 使溶解,作为对照药材溶液[5]。取除桂枝外的其他药材制成凝胶贴膏,按照上述方法制备桂枝阴性对照溶液。按照《中国药典》2020 年版的薄层色谱法,吸取上述三种溶液各20 μl,分别点于同一硅胶G 薄层板上,以石油醚(60~90 °C)-乙酸乙酯(17:3)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,且阴性无干扰,结果见图1D。
2.1.5 猪苓
取本品1 片,除去保护膜,称取药膏10 g,加甲醇100 ml,加热回流1 h,过滤,滤液蒸干,残渣加热水30 ml 使溶解,加三氯甲烷振摇提取2 次,每次30 ml,合并三氯甲烷液,蒸干,残渣加甲醇1.5 ml 使溶解,作为供试品溶液。另取麦角甾醇对照品,加甲醇制成每l ml 含1 mg 的溶液,作为对照品溶液。取除猪苓外的其他药材制成凝胶贴膏,按照上述方法制备猪苓阴性对照溶液。按照《中国药典》2020 年版的薄层色谱法,吸取供试品溶液、阴性对照溶液各20 μl、对照品溶液1 μl,分别点于同一硅胶G 薄层板上,以石油醚(60~90 °C)-乙酸乙酯(3∶1)为展开剂,展开,取出,晾干,喷以2%香草醛硫酸溶液,在105°C 加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,且阴性无干扰,结果见图1E。
2.1.6 预知子
取本品1 片,除去保护膜,称取药膏10 g,加甲醇100 ml,加热回流1 h,过滤,滤液挥干,残渣加热水20 ml 使溶解,加于D101 型大孔吸附树脂柱(内径为1.5 cm,柱高为12 cm)上,用水100 ml 洗脱,弃去水液,再用90%乙醇100 ml 洗脱,收集洗脱液,蒸干,残渣加甲醇2 ml 使溶解,作为供试品溶液。另取预知子对照药材1 g,精密称定,置具塞锥形瓶中,精密加入75%甲醇100 ml,密塞,称定重量,超声处理(功率300 W,频率50 kHz)30 min,放冷,再称定重量,用75%甲醇补足减失的重量,摇匀,滤过,取续滤液10 ml,蒸干,残渣加甲醇l ml 使溶解,作为对照药材溶液。取除预知子外的其他药材制成凝胶贴膏,按照上述方法制备预知子阴性对照溶液。按照《中国药典》2020 年版的薄层色谱法,吸取供试品溶液、阴性对照溶液各12 μl,对照药材溶液2 μl,分别点于同一硅胶G 薄层板上,以三氯甲烷-甲醇-水(13:4:1) 的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105°C 加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,且阴性无干扰,结果见图1F。
2.2 黄芪甲苷含量测定
2.2.1 色谱和质谱条件
色谱条件:色谱柱为ACQUITY UPLC®BEH C18柱(2.1 mm×100 mm,1.7 μm);流速:0.2 ml/min;柱温:40 ℃;进样量:1 μl;流动相:A 为含0.1%甲酸的乙腈溶液,B 为0.1%甲酸溶液,梯度洗脱(0~12 min,10%→90% A;12~13 min,90% A;13~13.5 min,90%→10% A;13.5~16 min,10% A)。
质谱条件:电喷雾离子(ESI)源,正离子检出模式,离子源温度:350 ℃,喷雾电压:3.5 kV,S-Lens RF 电压:50 V,毛细管温度:320 ℃,鞘气和辅助气均为高纯氮气(纯度>99.99%),鞘气压力:45 arb:辅助气压力:10 arb。数据采集采用一级全扫描(m/z783.47~787.47),分辨率:70 000。
2.2.2 对照品储备液的制备
精密称取黄芪甲苷对照品11.0 mg,置于100 ml量瓶中,加60%甲醇适量溶解并定容至刻度,摇匀,即得对照品储备液。精密量取黄芪甲苷对照品储备液25 ml,置于50 ml 量瓶中,用60%甲醇定容,即得黄芪甲苷对照品溶液(每1 ml 溶液含黄芪甲苷55 μg)。
2.2.3 供试品溶液的制备
揭去实脾消水凝胶贴膏覆膜和背衬材料,取膏体约5.0 g,精密称定,加入甲醇50 ml,称定重量,超声提取30 min,冷却到室温,再称定重量,用甲醇补足减失的重量,摇匀,用微孔滤膜(0.22 μm)滤过,即得。
2.2.4 阴性对照溶液的制备
按处方工艺制备不含黄芪的实脾消水凝胶贴膏阴性样品,按“2.2.3”项下方法制备阴性对照溶液。
2.2.5 方法专属性考察
分别吸取黄芪甲苷对照品储备液、供试品溶液和阴性对照溶液各1 μl,按“2.2.1”项下色谱和质谱条件进行测定。在正离子全扫描模式下黄芪甲苷色谱峰分离度良好,对照品及供试品溶液目标峰的保留时间一致,且阴性对照溶液无干扰(图2)。
2.2.6 线性关系考察
分别精密量取黄芪甲苷对照品溶液0.5、1、2、4、6 ml,置于10 ml 量瓶中,用60%甲醇定容至刻度,按“2.2.1”项下色谱和质谱条件进行测定,进样1 μl,记录峰面积。以黄芪甲苷的浓度为横坐标,峰面积为纵坐标,进行线性回归,得回归方程Y=0.967 7X+14.86,r=0.999 9,结果表明黄芪甲苷质量浓度在2.75~33 μg/ml 范围内与峰面积积分值呈良好的线性关系。
2.2.7 精密度试验
精密吸取同一对照品溶液1.0 μl,连续进样6 次,测定峰面积,结果黄芪甲苷峰面积的RSD 为2.42%,表明仪器精密度良好。
2.2.8 重复性试验
取批号为20180102 的样品6 份,精密称定,按“2.2.3”项下方法制备供试品溶液,按“2.2.1”项下条件进样测定峰面积,并计算含量,结果黄芪甲苷的含量分别为10.15、10.67、10.56、10.28、10.46、10.39 μg/g,RSD 为1.81%,表明该方法重复性良好。
2.2.9 稳定性试验
取“重复性试验”项下其中一份供试品溶液,于0、2、4、6、8、12、24 h 分别进样测定峰面积,结果黄芪甲苷峰面积的RSD 为2.53%,表明供试品溶液在24 h 内稳定性良好。
2.2.10 加样回收率试验
取实脾消水凝胶贴膏(批号:20180102)6 份,每份约2.5 g,精密称定,分别精密加入黄芪甲苷对照品溶液(5.5 μg/ml)1 ml,按“2.2.3”项下方法制备供试品溶液,按“2.2.1”项下色谱条件进样测定峰面积,并计算回收率,结果平均回收率为102.79%,RSD 为2.22%。
2.2.11 样品测定
分别取3 批次实脾消水凝胶贴膏,按照“2.2.3”项下方法制备供试品溶液,按“2.2.1”项下色谱和质谱条件检测,测定峰面积,计算黄芪甲苷的含量,结果见表1。
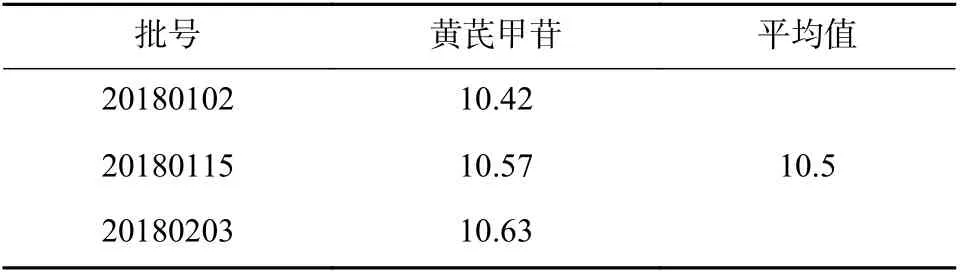
表1 黄芪甲苷含量测定结果(μg/g,n=3)
3 讨论
在对实脾消水凝胶贴膏进行薄层鉴别的过程中,由于凝胶贴膏中含有水溶性高分子化合物,如果用水提取,膏体就会吸水膨胀变粘,因此,本文选择甲醇作为供试品前处理提取溶剂。另外,为了富集特征化学成分,一些极性不大的化学成分可以采用萃取的方法,一些极性较大的化学成分可以采用大孔树脂柱层析的方法。本实验中笔者发现,10%乙醇[6]、30%乙醇[7]、60%乙醇[8]、90%乙醇大孔树脂洗脱液中分别含有红花、车前子、桃仁、预知子的特征成分,但是利用薄层色谱法进行鉴别时,红花的分离效果较差,桃仁阴性有干扰,故本文未将红花及桃仁列入质量标准。
虽然蒸发光散射检测器在那些弱或者无紫外吸收的中药成分分析中具有独特而不可替代的作用(例如黄芪甲苷的测定),但是蒸发光散射检测器在分析中药成分时还有许多不尽人意的地方[9]。课题组前期实验时也试过蒸发光散射检测器,为了使黄芪甲苷分离效果好,其保留时间需达到44 min左右,而且重现性差。而采用UPLC-MS 的方法,黄芪甲苷在10 min 内即可测定完,且分离效果好,故UPLC-MS 在不影响分离效果的情况下大大提高了样品中各成分的分析速度[10-11]。另外,在色谱和质谱条件的优化中,本实验考察了不同比例流动相(0.1%甲酸的乙腈-0.1%甲酸溶液)的分离效果,最终确定梯度洗脱的方式。因此,本研究采用UPLC-MS 的方法测定实脾消水凝胶贴膏中黄芪甲苷的含量,为实脾消水凝胶贴膏的质量控制提供了一种快速、灵敏、稳定、可靠的方法。
实脾消水浸膏为传统浸膏剂,处方有十二味药,其质量标准除了常规检查项外,主要是【鉴别】项,要求对黄芪、牵牛子、车前子、桂枝和冰片进行薄层色谱鉴别。本文建立了实脾消水凝胶贴膏的质量控制方法,在原标准的基础上增加了【含量测定】项,并在【鉴别】项下增加了莪术、猪苓、预知子的薄层色谱鉴别,为实脾消水浸膏的二次开发奠定了一定的基础。
猜你喜欢
老年博览·上半月(2020年10期)2020-10-21
中成药(2018年5期)2018-06-06
临床医药文献杂志(电子版)(2017年11期)2017-05-17
海峡科技与产业(2016年3期)2016-05-17
云南中医学院学报(2015年3期)2015-07-31
西部中医药(2015年9期)2015-02-02
安徽医药(2014年4期)2014-03-20
中国合理用药探索(2014年11期)2014-03-11
中成药(2014年8期)2014-02-28
湖南中医药大学学报(2011年7期)2011-03-20
