
基于CRISPR/Cas9技术验证人T淋巴细胞中单等位突变型UNC13D基因脱颗粒功能
2023-12-01 12:22:10蔡丽莎徐晓军
遵义医科大学学报 2023年11期
蔡丽莎,邢 源,黄 佩,陈 艳 ,徐晓军
(1.遵义医科大学附属医院 小儿内科,贵州 遵义 563099;2.浙江大学医学院附属儿童医院 血液肿瘤内科,浙江 杭州 310003;3.贵州省儿童医院 小儿内科,贵州 遵义 563099;4.遵义医科大学 组织损伤修复与再生医学省部共建协同创新中心,贵州 遵义 563099)
噬血细胞性淋巴组织细胞增多症(hemophagocytic lymphohistiocytosis, HLH)是一种由于机体内单核巨噬细胞以及淋巴细胞过度激活,从而产生大量炎症因子的高炎症反应疾病[1-2],死亡率高,成人及儿童均可见[3]。家族性噬血细胞性淋巴组织细胞增多症(familial hemophagocytic lymphohistiocytosis, FHL) 是由参与颗粒依赖性胞吐作用途径的基因突变引起的,其相关基因突变会导致自然杀伤(nature kill, NK)细胞和T淋巴细胞的细胞毒活性受损[4]。现已发现有10余种基因的突变与 HLH 的发病明确相关,如PRF1、UNC13D、STX11、STXBP2、RARB27A、LYST、AP3B1、SH2D1A和BIRC4 等[5]。FHL是一种常染色体隐性遗传病,这意味着只有纯合子或复合杂合子变异才具有致病可能性,而杂合子携带者则不表现出临床症状。然而,一些报道已经介绍了有症状的杂合变异携带者[6],并且随着全基因组测序的开展,越来越多的单等位基因杂合突变在HLH患者中被发现[7],但是其具体位点突变所造成的功能影响却鲜有报道。编码Munc13-4蛋白的基因UNC13D突变会引起家族性噬血细胞性淋巴组织细胞增生症3型(FHL3)[8],FHL3几乎占FHL发病的30%~40%[9]。CRISPR/Cas9基因编辑系统,由于其高效率和高精度,在血液病、肿瘤和其他遗传疾病领域都展现出极大的应用前景[10]。本研究通过CRISPR/Cas9 技术敲除人原代T淋巴细胞中的UNC13D基因,经抗生素筛选并扩增出Munc13-4 蛋白极低表达的T淋巴细胞进行体外扩增,再将前期已包装好的野生型以及突变型UNC13D(c.1232 G>A, p.R411Q)慢病毒感染knockout-UNC13D的人T淋巴细胞中,建立基于CRISPR/Cas9基因编辑重建技术检测HLH相关基因UNC13D单等位基因突变的致病性验证方法,以此来判断该UNC13D基因突变位点是否具有致病性。
1 材料与方法
1.1 主要试剂及仪器 人胚肾293T细胞株(美国ATCC);质粒Lenti CRISPR V2(美国Addgene公司);野生型和突变型(c.1232 G>A, p.R411Q)pGV348-UNC13D慢病毒(保存于本实验室中);IMDM、RPMI-1640培养基、胎牛血清(美国Gibco公司);X-VIVO培养基(德国LONZA公司);人淋巴细胞分离液(天津灏洋生物公司);CD3/CD28磁珠(德国Miltenyi公司);人IL-2(美国PeproTech公司);限制性内切酶BsmBI酶、T7核酸内切酶和T4连接酶(美国NEB公司);感受态Stbl3(上海昂羽生物技术公司);促转剂Polybrene、高保真酶、脂质体转染试剂(上海YEASEN公司);病毒滴度检测试剂(日本 TAKARA 公司);转染级质粒提抽试剂盒(德国QIAGEN公司);基因组抽提试剂盒(北京天根生物公司);DiaSpin柱式DNA胶回收试剂盒(上海生工生物公司);鼠抗人CD3-BV421、CD107a-PE、CD8-PE-Cy7抗体(美国Biolegend公司);兔抗人FLAG-PE抗体、一抗兔抗人Munc13-4抗体(英国Abcam公司);二抗PE标记的驴抗兔抗体(美国invitrogen公司)。普通PCR仪、低温高速离心机(均为美国Thermo公司);水平电泳仪(美国BioRed公司);凝胶成像仪(美国GE公司);酶标仪(美国BioTek公司)。实验相关引物合成以及测序均委托杭州有康生物科技公司完成。
1.2 方法
1.2.1 gRNA靶位点的选择 针对目的基因靶基因序列,利用公用网站(CHOPCHOP网站)中提供的gRNA序列设计原则,设计多个靶位点序列,根据设计软件进行评估测定,选择最佳的动力学参数靶点,最后在UNC13D基因的第6个外显子处设计出第一条单靶点sgRNA1 (图1A)、两对双靶点的sgRNA2-1、sgRNA2-2、sgRNA3-1以及sgRNA3-2(图1B),序列见表1、2。

表1 sgRNA1 oligo序列

表2 双靶sgRNA2和sgRNA3序列
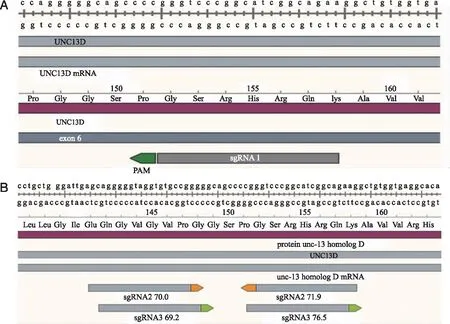
A: sgRNA1 ;B: sgRNA2和sgRNA3。图1 设计的单靶点sgRNA1与双靶点sgRNA2和sgRNA3
1.2.2 CRISPR/Cas9 UNC13D-sgRNA重组质粒的构建 首先用BsmBI酶切lentiCRISPR V2(CRISPR/Cas9)载体与退火后的sgRNA混合,T4 DNA连接酶在25 ℃水浴中孵育30 min,然后将连接产物转化至Stbl3感受态细胞中,涂平板,用氨苄抗性筛选阳性克隆,挑取单克隆,提取质粒并测序鉴定。
1.2.3 转染及验证UNC13D基因的敲除情况 重组质粒转染293T细胞,48 h后流式检测转染效率并提取293T细胞基因组DNA,对目标区域6号外显子进行PCR,引物见表3;将 PCR产物进行琼脂糖凝胶电泳检测,回收目的DNA片段并送有康生物公司进行Sanger测序;同时用 T7 核酸内切酶检测切割效率,最后选择切割效率较高的sgRNA序列。
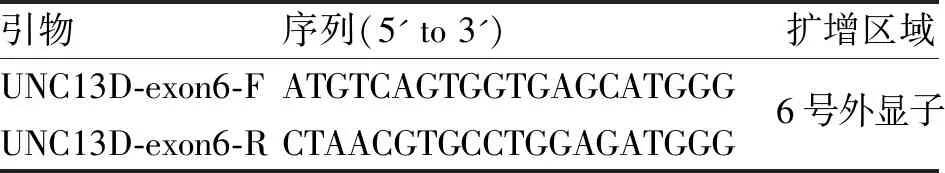
表3 UNC13D基因6号外显子扩增引物序列
1.2.4 慢病毒包装与滴度检测 使用慢病毒二代包装系统(三质粒包装系统)、目的质粒CRISPR/Cas9-sgRNA2、慢病毒的包装质粒psPAX2以及pMD2.G。前1 d将293T细胞接种于10 cm细胞培养皿中,当细胞密度接近70%~80%时,使用脂质体转染试剂进行转染,分别在转染24 h以及52 h后,收集病毒上清,用0.45 μm滤膜过滤后,加入慢病毒浓缩试剂后4 ℃过夜,用适宜体积的IMDM培养基重悬病毒。使用慢病毒滴度试剂盒检测慢病毒滴度,分装后保存于-80 ℃冰箱。
1.2.5 CRISPR/Cas9 UNC13D-sgRNA慢病毒感染人T淋巴细胞并检测慢病毒感染效率 先分离出健康志愿者的外周血单个核细胞(PBMC),用CD3/CD28磁珠以及人白介素2(interleukin,IL2)激活T淋巴细胞,3 d后取 T淋巴细胞3.5×105个,按MOI=100加入CRISPR/Cas9 UNC13D-sgRNA慢病毒,并加入Polybrene至终浓度为8 μg /mL,700 g水平离心2 h,放入37 ℃、5%CO2培养箱孵育,24 h后更换为X-VIVO完全培养基。感染3 d后流式检测慢病毒感染效率,T淋巴细胞先加表面抗体CD3以及CD45各3 μL室温避光孵育30 min,PBS洗涤后进行固定破膜,然后加入FLAG-PE抗体3 μL,对照管加入同型抗体3 μL,室温避光孵育30 min,PBS洗涤2次后重悬细胞上机检测。
1.2.6 野生型和突变型UNC13D慢病毒再次感染人T淋巴细胞 CRISPR/Cas9 UNC13D-sgRNA慢病毒感染人T淋巴细胞3 d后加入嘌呤霉素至终浓度为1 μg/mL,放于细胞孵箱中培养24 h后更换为完全培养基,再继续培养3~5 d后将人T淋巴细胞分两组后分别加入实验室已保存好的野生型(Wt)和突变型(Mut, c.1232 G>A, p.R411Q)pGV348-UNC13D慢病毒再次感染knockout-UNC13D基因的人T淋巴细胞。
1.2.7 流式细胞术检测人T淋巴细胞Munc13-4蛋白表达情况 CRISPR/Cas9 UNC13D-sgRNA慢病毒感染人T淋巴细胞3 d后以及嘌呤霉素药筛24 h后分别检测Munc13-4蛋白的表达。取T淋巴细胞4×105个,加入表面抗体CD3以及CD45各3 μL室温避光孵育30 min,PBS洗涤1次后进行固定破膜,然后加入一抗兔抗人Munc13-4抗体5 μL,对照管加入同型抗体5 μL,室温避光孵育30 min后PBS洗涤1次后再加入二抗PE标记的驴抗兔抗体2 μL,PBS洗涤1次后重悬细胞上机检测。野生型(Wt组)和突变型UNC13D(Mut组)慢病毒再次感染人T淋巴细胞3 d后同样采用流式细胞术检测T淋巴细胞Munc13-4蛋白表达。
1.2.8 流式细胞术检测CTL的脱颗粒功能 CRISPR/cas9 UNC13D-sgRNA慢病毒感染人T淋巴细胞,经嘌呤霉素药筛后流式细胞术检测细胞毒性T淋巴细胞(cytotoxic T lymphocyte,CTL)的脱颗粒功能。取T淋巴细胞3×105个于48孔板中培养,加入2 μL CD107a-PE抗体以及1 μL CD3/CD28磁珠刺激,阴性对照管只加入2 μL CD107a-PE抗体,同型对照管加入同型抗体2 μL,放入37 ℃、5% CO2的细胞培养箱中培养4 h;PBS洗涤1次后加入CD3-BV421以及CD8-PE-Cy7抗体各3 μL,室温下避光孵育30 min,PBS洗涤1次后重悬细胞上机检测;野生型(Wt组)和突变型UNC13D(Mut组)慢病毒再次感染人T淋巴细胞3 d后同样采用流式细胞术检测CTL的脱颗粒功能。脱颗粒功能ΔCD107a=刺激组的CD107a-非刺激组的CD107a。
2 结果
2.1 CRISPR/Cas9 UNC13D-sgRNA重组质粒的构建与鉴定 凝胶电泳显示LentiCRISPR V2载体BsmBⅠ酶切成功(图2),分别将两条互补的sgRNA退火形成双链后连接到用酶切后的CRISPR/Cas9载体中,测序结果显示3对sgRNA成功连接至CRISPR/Cas9载体中。
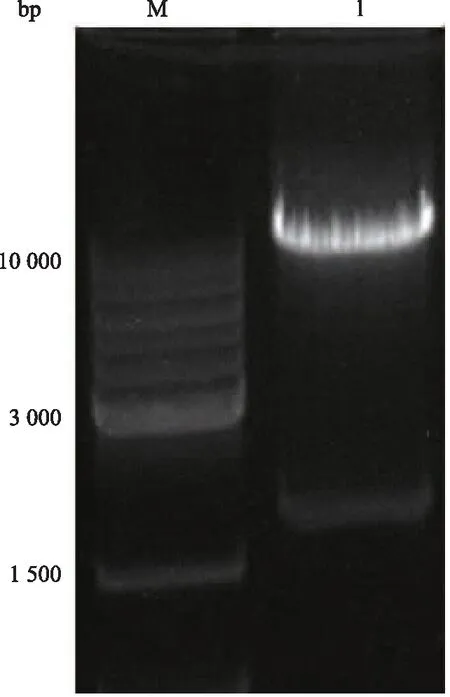
图2 LentiCRISPR V2酶切后电泳结果
2.2 293T细胞转染效率的检测 将构建好的3种CRISPR/Cas9 UNC13D-sgRNA分别转染到293T细胞中,流式细胞术检测293T细胞的转染效率,结果显示质粒成功转入293T细胞中,感染效率在10%~20%(图3)。

图3 流式检测293T细胞的转染效率
2.3 基因测序及T7酶切法验证sgRNA的敲除能力 将3种sgRNA重组质粒瞬转293T细胞并抽提细胞基因组DNA,以基因组DNA为模板对目的序列进行PCR扩增(图4A)。将一部分纯化后的PCR产物经T7酶进行酶切凝胶电泳验证出这3对sgRNA对UNC13D基因均具有切割能力,阴性对照组无切割能力(图4B)同时将纯化后的PCR产物送有康生物公司进行Sanger测序,从测序峰图可见位于 sgRNA 位置附近的 DNA 序列测序峰图出现明显的杂乱无章的重叠峰图,而阴性对照组的测序结果无重叠峰图的出现(图5)。

A: PCR扩增电泳;B: 扩增产物T7酶切电泳(1、2、3泳道分别为sgRNA1、2、3、4泳道为Control)。图4 293T细胞基因组DNA进行PCR扩增以及T7酶切电泳结果
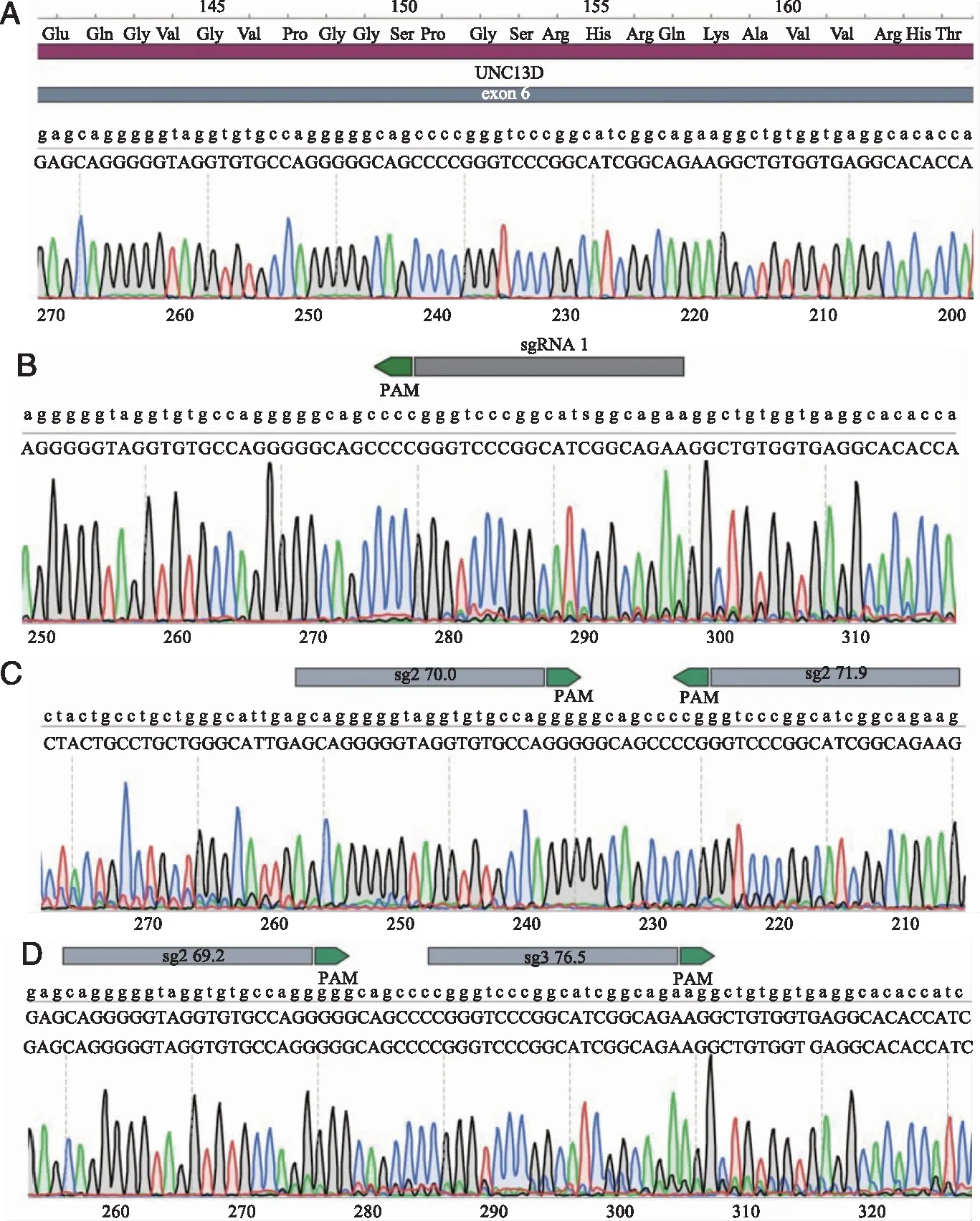
A: 空载LentiCRISPR V2;B:sgRNA1;C: sgRNA2;D: sgRNA3。图5 PCR扩增产物测序结果
2.4 流式细胞仪检测慢病毒感染T淋巴细胞的转染效率 本研究选择CRISPR/Cas9-control和CRISPR/Cas9-sgRNA2包装慢病毒、浓缩以及滴度测定后感染人T淋巴细胞,PBMC经过CD3/CD28磁珠刺激活化后CD3+T淋巴细胞比例高达98%,CTL比例为41.4%(图6A),感染72 h后测定慢病毒的感染率,流式结果显示两种慢病毒感染T淋巴细胞的感染率在30%~40%(图6B),可以用作后续实验。
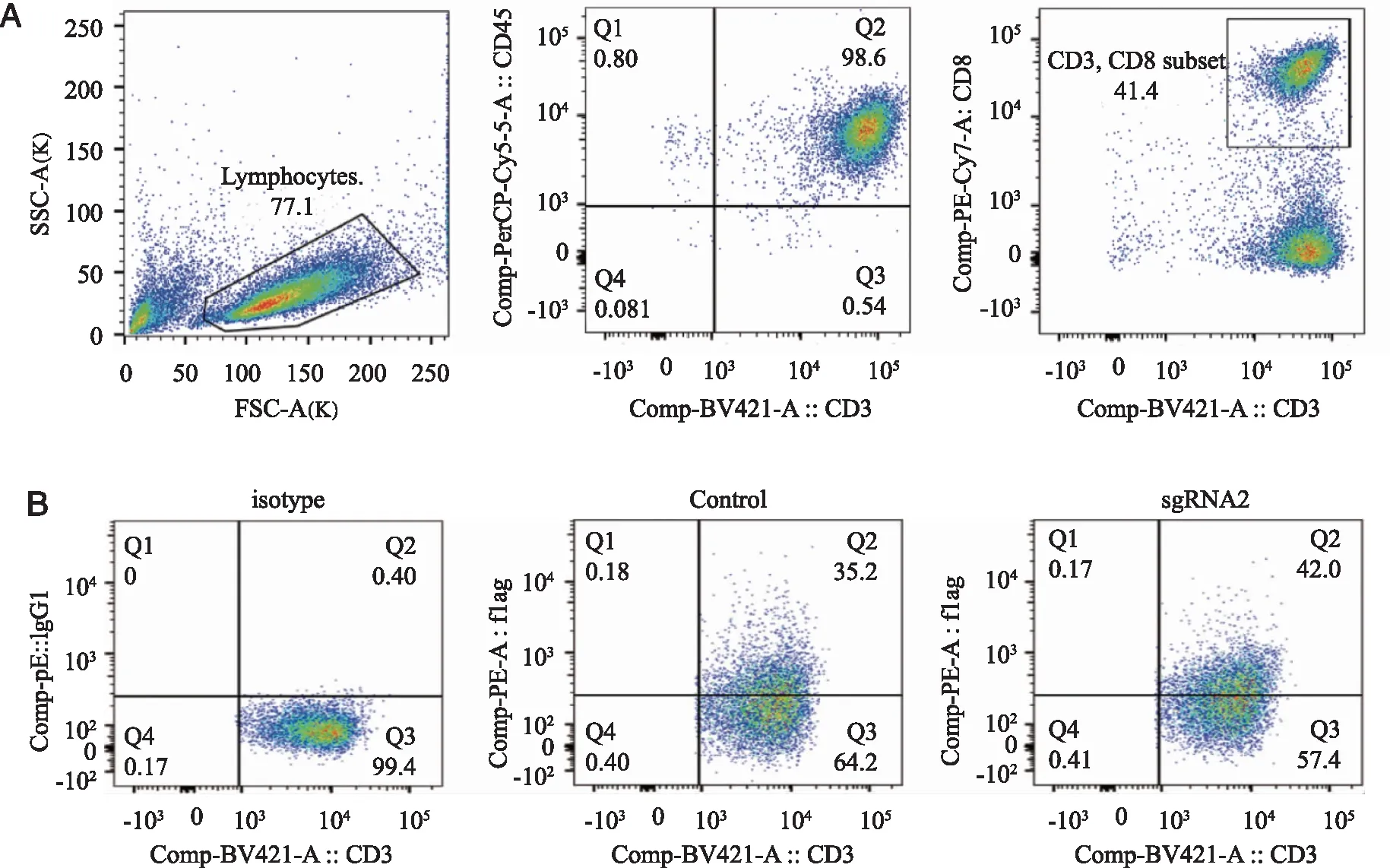
A: PBMC活化后T淋巴细胞比例以及CTL的比例;B: Control和sgRNA2慢病毒的感染效率。图6 慢病毒感染T淋巴细胞72 h后感染率
2.5 流式细胞仪检测慢病毒感染T淋巴细胞内蛋白表达 与感染空载慢病毒的Control组相比,感染sgRNA2的T淋巴细胞胞内Munc13-4蛋白表达下降,Control组为51.8%,sgRNA2组为34.9%。加入嘌呤霉素药筛24 h后再次检测T淋巴细胞胞内Munc13-4蛋白的表达,结果显示感染sgRNA2慢病毒的T细胞内Munc13-4蛋白显著降低,Control组为33.6%,sgRNA2组为3.41%(图7A),表明UNC13D基因成功被敲低。
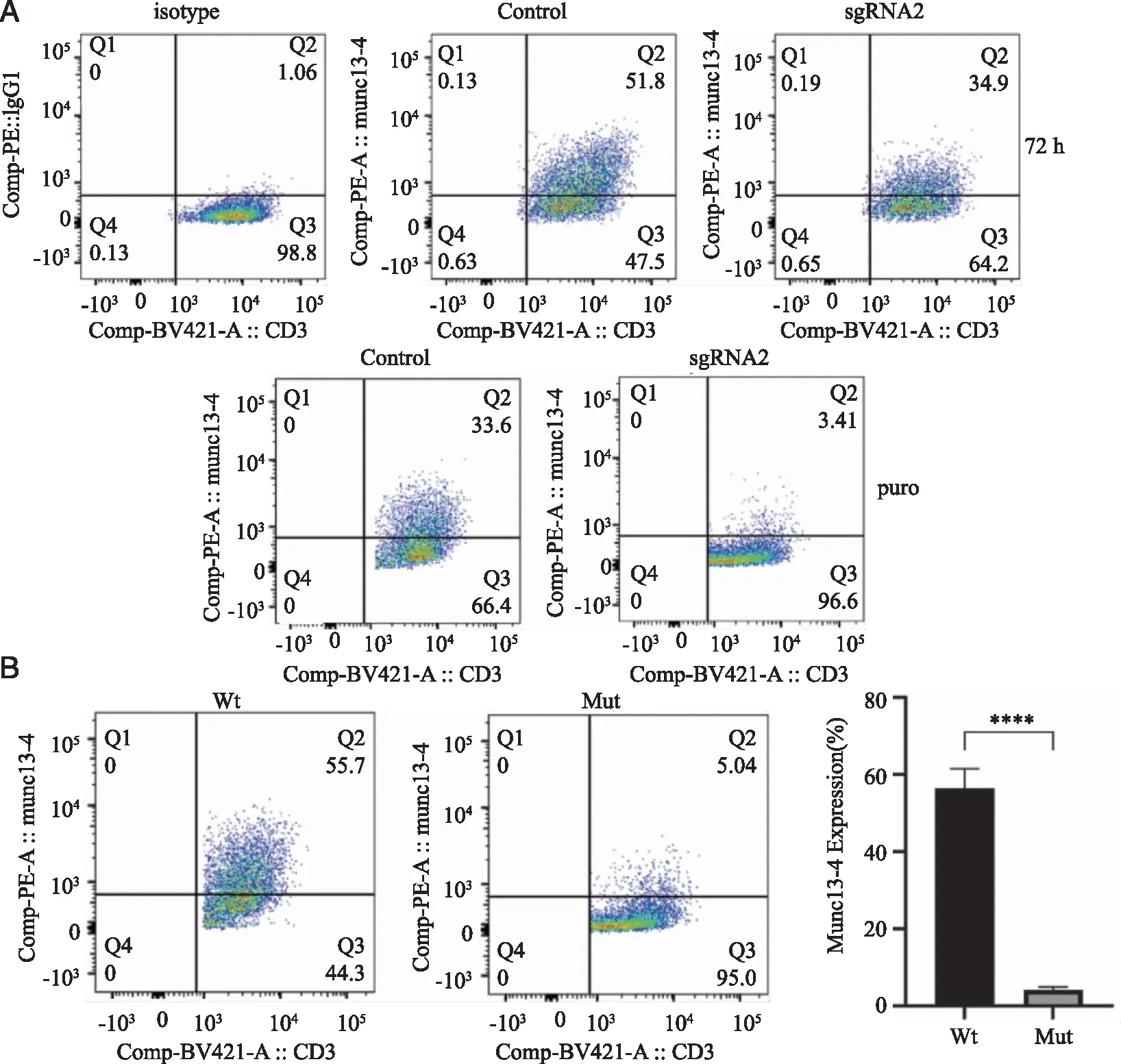
A:慢病毒感染后72 h以及puro筛选后蛋白的表达;B:Wt和Mut感染敲除后的T淋巴细胞中蛋白的表达;****:P<0.000 1。图7 流式检测T淋巴细胞内Munc13-4蛋白表达
再将实验室前期保存好的野生型(Wt)以及突变型(Mut)UNC13D慢病毒再次感染knockout-UNC13D基因的T淋巴细胞中,感染3 d后流式检测T淋巴细胞内Munc13-4蛋白的表达,结果显示Wt组的T淋巴细胞内Munc13-4蛋白较敲除后升高,但Mut组的T细胞内Munc13-4蛋白较敲除后无明显变化(图7B,P<0.000 1),表明该突变型UNC13D基因无法编码Munc13-4蛋白。
2.6 流式细胞仪检测CTL脱颗粒功能 与感染空载慢病毒的Control组相比,感染sgRNA2的CTL中的ΔCD107a明显减弱,脱颗粒Control组:45.1%;sgRNA2组:11.4%(图8A),表明敲除UNC13D基因后,CTL的脱颗粒功能明显下降。

A: UNC13D基因敲除后对CTL脱颗粒功能的影响;B: Mut对敲除后CTL脱颗粒功能的影响;***:P<0.001。图8 流式细胞术检测CTL的脱颗粒功能
再将实验室前期保存好的野生型(Wt)以及突变型(Mut)UNC13D慢病毒感染knockout-UNC13D基因的T淋巴细胞中,感染3 d后流式细胞术再次检测CTL的脱颗粒功能,结果显示Wt组的CTL中的ΔCD107a较敲除后有所升高,但Mut组的CTL中的ΔCD107a较敲除后无明显变化(图8B,P<0.001),表明该突变型UNC13D基因能够使人CTL脱颗粒功能下降。
3 讨论
家族性噬血细胞性淋巴组织细胞增多症(FHL)是由细胞毒性T淋巴细胞(CTL)和NK细胞的细胞毒性活性异常引起的先天性免疫缺陷疾病,CTL和巨噬细胞过度活化会导致高细胞因子血症而引发多器官功能衰竭[11]。家族性噬血细胞性淋巴组织细胞增生症3型 (FHL3)是由编码Munc13-4蛋白的UNC13D基因突变所引起的,该基因定位于染色体17q25,包含了32个外显子,共编码1 090个氨基酸[8]。FHL是常染色隐性遗传病,理论上杂合子携带者并不会发病,但是随着全基因组测序的开展,越来越多的文献报道HLH 患者中检测出了单等位基因的杂合突变[7, 12];因此,目前需要建立一种判断单等位基因杂合突变是否会引起HLH发病的验证方法。
UNC13D基因编码与细胞毒性淋巴细胞中对裂解颗粒的转运和胞吐作用有关的蛋白质Munc13-4,定量测定该蛋白有助于 HLH 患者的诊断[13]。CD107a是一种溶酶体蛋白,能与CTL和NK细胞的裂解颗粒中的穿孔素共定位;T细胞抗原受体(T cell receptor, TCR)与CTL的结合以及NK细胞上的一系列活化受体可以诱导CD107a的表面表达,能够反映裂解颗粒的胞吐作用[14-15]。目前有文献报道从人外周血中分离出单个核细胞(PBMC),并通过CD3/CD28磁珠与IL-2、IL-7、IL-15等细胞因子结合离体激活并扩增T淋巴细胞,再将慢病毒转导入活化后的T淋巴细胞中的技术方法[16-17]。本研究用CD3/CD28磁珠以及IL-2刺激并活化T淋巴细胞,再将慢病毒导入活化后的T淋巴细胞中来进行实验。有研究报道通过K562细胞刺激NK细胞以及通过CD3/CD28磁珠刺激CTL来检测其脱颗粒功能的方法[14,18],本课题组前期也建立了CD3/CD28磁珠刺激CTL并采用流式细胞术检测脱颗粒的方法来验证CTL的细胞毒功能[19],同时已构建好野生型与突变型UNC13D基因慢病毒表达载体(突变位点为14号外显子点突变c.1232 G>A,p.R411Q)并成功包装好慢病毒。
CRISPR/Cas9是一种由RNA介导的DNA内切酶,能够通过与靶序列互补的单诱导RNA(single guide RNA, sgRNA)诱导至基因组中[10, 20],当CRISPR/ Cas9 在sgRNA诱导至基因组后,它将会与靶序列结合,然后在sgRNA附近的基因组区域产生DNA的双链断裂。由于双链断裂后的DNA可以通过同源重组修复或非同源重组修复,最终引起碱基的突变、插入或缺失,从而可达到特异性基因编辑的目的[21],在利用CRISPR-Cas9系统进行基因编辑时,可以应用脂质体、纳米材料或慢病毒作为递送系统,将Cas9以及sgRNA导入基因组中[22-23]。并且CRISPR/Cas9基因编辑技术在癌症免疫治疗中的应用也十分广泛,其介导的基因组编辑可用于消除编码抑制性T细胞表面受体的基因,例如程序性细胞死亡蛋白1(programmed cell death proteinl,PD-1)和细胞毒性T淋巴细胞相关蛋白4(cytotoxic T lymphocgte-associatd antigen-4,CTLA-4),以提高基于T细胞的癌症免疫治疗的效率[24]。
目前已经有研究采用过表达法检测HLH相关单等位基因突变后NK细胞脱颗粒功能的方法[25-26],但是由于正常背景的存在下,NK细胞与CTL细胞毒功能下降的量化无法明确。本研究采用CRISPR/Cas9基因编辑技术来敲除人T淋巴细胞中的UNC13D基因,从而消除正常背景所带来的影响。本实验中慢病毒感染人T淋巴细胞效率在30%~40%,因此通过嘌呤霉素药筛24 h后以增加knockout-UNC13D基因的T淋巴细胞的比例,CRISPR/Cas9-sgRNA慢病毒感染人T淋巴细胞后Munc13-4蛋白的表达以及CTL脱颗粒功能明显下降,表明成功敲除人T淋巴细胞中的UNC13D基因。再将野生型以及突变型UNC13D慢病毒感染knockout-UNC13D基因的人T淋巴细胞后,结果表明位点c.1232 G>A(p.R411Q)的突变型UNC13D基因能够使CTL脱颗粒和细胞毒性活性降低。
综上所述,本研究利用CRISPR/Cas9基因编辑技术敲除人T淋巴细胞中UNC13D基因,下调Munc13-4蛋白的表达,验证出UNC13D基因的突变位点c.1232 G>A(p.R411Q)为致病性突变位点。成功建立基于CRISPR/cas9基因编辑重建技术检测HLH相关基因UNC13D单等位基因突变的致病性验证方法,为研究FHL的机制提供了新方向。但本研究还有不足之处,由于原代T淋巴细胞无法进行亚克隆筛选,因此未能建立完全敲除UNC13D基因的T淋巴细胞,以及未建立出UNC13D基因敲除的模型鼠,后续还将进一步完善该实验技术。
猜你喜欢
中老年保健(2022年1期)2022-08-17 06:14:30
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
兽医导刊(2019年1期)2019-02-21 01:14:20
猪业科学(2018年8期)2018-09-28 01:27:52
现代检验医学杂志(2015年6期)2015-02-06 01:44:07
安徽医药(2014年9期)2014-03-20 13:14:09
吉林大学学报(医学版)(2014年2期)2014-02-27 06:48:05
安徽医科大学学报(2014年11期)2014-02-15 10:20:36
现代检验医学杂志(2014年3期)2014-02-02 02:42:05
现代检验医学杂志(2014年2期)2014-02-02 02:40:39
