
血管新生过程中的Rho GTPase
2023-11-24 07:34:06施伟丽高海霞吴鸿
医学分子生物学杂志 2023年6期
施伟丽 ,高海霞 ,吴鸿
1河南中医药大学第二临床医学院 郑州市,450002
2河南省中医院 郑州市,450002
缺血性心脑血管疾病(包括缺血性心脏病、缺血性卒中) 依然是中国城乡居民死亡的首要原因,给个人、家庭和社会带来沉重的负担[1]。新生血管不足是导致心脑血管循环障碍和组织坏死的主要原因之一,促进缺血组织血管新生是缺血性心脑血管疾病治疗的有效手段。血管新生是在原有毛细血管和/或微静脉基础上,以芽生或非芽生的形式生成新的毛细血管的过程,其中内皮细胞贯穿血管新生始终。Rho 家族的鸟苷酸三磷酸酶(Rho GTPase) 通过与二磷酸鸟苷(guanosine diphosphate,GDP) 结合的非活性形式或与三磷酸鸟苷(guanosine triphosphate,GTP) 结合的活性形式的相互转换实现其在多种病理和生理过程中的分子开关作用,参与血管新生过程,其机制包括调节肌动蛋白细胞骨架影响内皮细胞迁移、通过黏着斑调节细胞黏附和连接,并在生长因子等信号影响下调节内皮细胞成管等。本文旨在深入探讨Rho GTPase在血管新生中的作用为缺血性心脑血管疾病的治疗提供思路。
1 血管新生的过程
血管新生涉及细胞外基质降解、内皮细胞特化和对促血管新生信号的识别、内皮细胞迁移等生物过程。在健康成年人中,静止的内皮细胞半衰期较长,这有利于内皮维持自身的自分泌作用。在缺血缺氧损伤等应激状态下,为建立有效的灌注腔并防止内皮细胞盲目沿血管生成信号的方向移动,位于新生血管最前端的单个、高度极化且具有特殊形态和功能的部分细胞由休眠状态转变为激活状态。即为内皮尖细胞。内皮尖细胞是血管生成过程中位于血管芽最尖端且具有特殊形态和功能的内皮细胞,可伸出富含肌动蛋白且长短不一的丝状伪足,丝状伪足向前延伸,能充分感知周围环境中各种促血管生长的因子,如血管内皮生长因子(vascular endothelial growth factor,VEGF),从而引导和控制内皮细胞迁移方向。此外,丝状伪足通过折叠和黏附内皮细胞钙黏蛋白,开启细胞间接触和内皮细胞间的桥接。丝状伪足(filopodia) 后缘紧邻片状伪足(lamellipodia),片状伪足是细胞迁移过程中前向运动的马达。单个内皮细胞迁移包括细胞前沿丝状伪足和片状伪足形成、延长并与细胞外基质黏附形成黏着斑与新的牵拉点。
2 Rho GTPase 家族
Rho GTPase 是小G 蛋白的一种,哺乳动物Rho 家族按其氨基酸结构分为8 个亚家族[2]。Rho GTPase 具有GTP 酶活性,通过非活性GDP 结合形式和活性GTP 结合形式实现其在多种病理和生理过程中的分子开关作用。其中,鸟苷酸交换因子(nucleotide exchange factors,GEFs) 促进GTPase与GTP 结合,从而激活下游底物进行信号转导,只有和质膜偶联的小GTPase 才能被GEF 激活(GEF 包括DBL 和DOCK 家族);而GTP 酶活化蛋白(GTPase-activating proteins,GAPs) 增加Rho GTPase 分子内部GTP 水解,使Rho GTPase 失活而关闭信号转导;GDP 解离抑制因子(guanosine nucleotide dissociation inhibitors,GDIs) 抑制GDP 从Rho GTPase 上解离。大多数小GTPases 在C 端存在脂质修饰,以便锚定在细胞膜上,同时GDIs 通过脂类基团与GTPases 结合。此外,Rho GTPase 活性也受翻译后修饰影响,如磷酸化、泛素化等。Rho家族蛋白参与细胞骨架重组、细胞黏附和迁移等过程,并在血管新生中发挥重要作用。
3 Rho GTPase 家族成员参与血管新生
细胞分裂周期蛋白(cell division cycle 42,Cdc42)、Rac1 和RhoA 是Rho GTPase 家族中参与血管新生被研究最多的因子,他们参与血管新生的机制包括调节肌动蛋白细胞骨架、影响内皮细胞迁移、通过黏着斑调节细胞黏附和连接并在生长因子等信号影响下调节内皮细胞成管等(图1)。
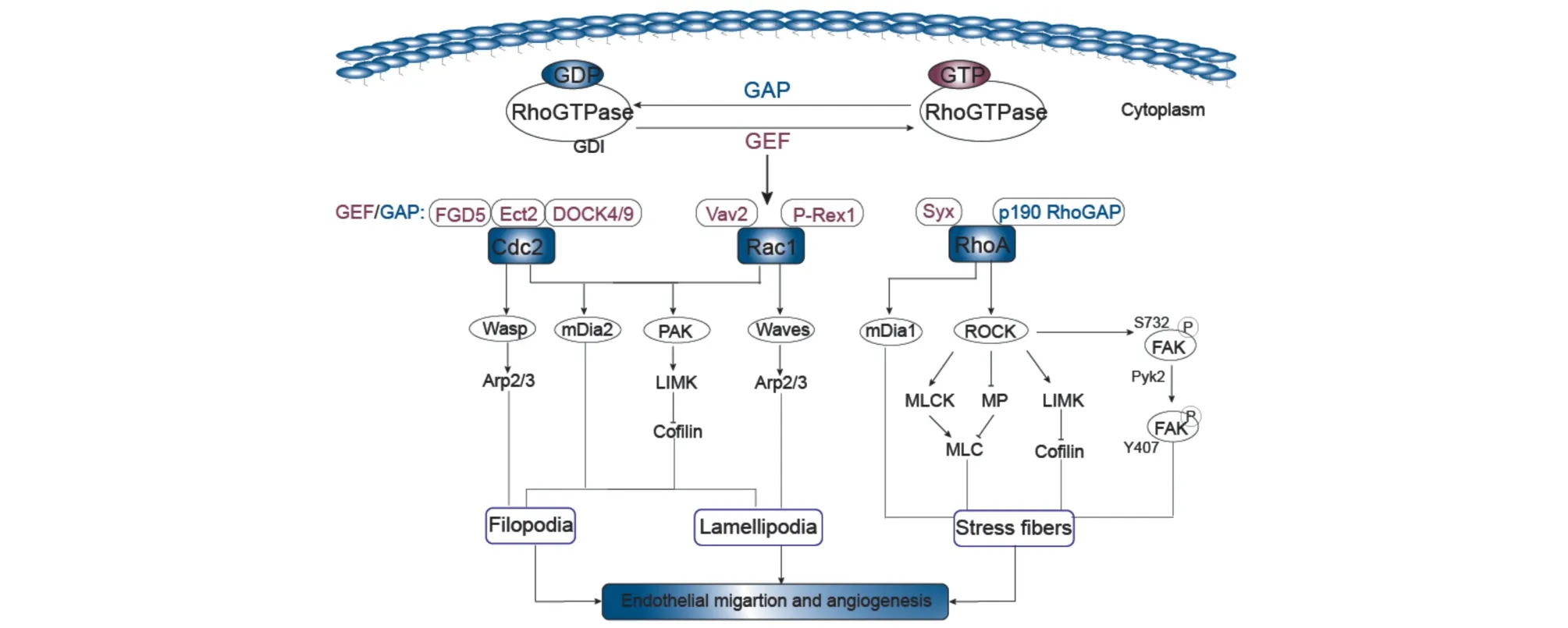
图1 血管新生过程中的Rho GTPase
3.1 Cdc42
正确的迁移方向是内皮细胞形成有效血管腔的基础,Cdc42 区域性的激活引起微管和微丝等细胞骨架的极性分布,从而指引内皮细胞的爬行方向。诱导细胞迁移的外界信号,如VEGF/NRP1 通过GEF 区域性地激活Cdc42,GTP-Cdc42 聚集到细胞迁移的前端,通过WASP-Arp2/3 促进内皮尖细胞丝状伪足的形成,进而刺激细胞骨架微丝的形成,引导自由悬浮的Cdc42 聚集到细胞膜上,细胞膜某部分Cdc42 浓度升高,诱导内皮尖细胞迁移和出芽[3-4]。内皮特异性敲除Cdc42 的小鼠由于血管生成缺陷和微血管通透性增加而具有胚胎致死性[5],可见Cdc42 对血管形成有重要作用。活化的Cdc42可能通过结合和激活成蛋白样蛋白(formin-like protein 3,Fmnl3) 促进内皮细胞丝状伪足的形成和延伸,从而诱导斑马鱼尾静脉丛血管出芽[6]。此外,Cdc42 参与调节血管内皮细胞屏障的恢复,其激活有利于内皮细胞紧密连接的形成,从而恢复其完整性[7]。Pannekoek等[8]发现cAMP-Epac1 通路激活Rac1 和Rap1,招募FGD5(Rho GTPase 的GEF) 到细胞连接处,活化Cdc42 从而稳定内皮细胞钙黏蛋白带,促进黏合连接重组装从而恢复内皮屏障功能。Cdc42 的GEF Ect2 与紧密连接衔接蛋白Par3-Par6-aPKC(PAR 蛋白复合物) 结合,激活Cdc42/aPKC 参与新生血管内皮屏障的形成[9]。
Cdc42 对血管新生的调节功能离不开其与生长因子的交互作用(表1)。当内皮细胞Cdc42 特异性缺失时,血管内皮生长因子受体2(vascular endothelial growth factor receptor 2,VEGFR2) 的片段化增加,导致内皮细胞迁移功能减弱[10]。Cdc42的GEF DOCK4 通过与DOCK9 的相互作用控制内皮细胞丝状足突的形成,这对内皮细胞侧枝重塑和管腔形态维持是必须的,而VEGF 促进DOCK4 与DOCK9 的相互作用[11]。研究发现,Cdc42 亚型RhoJ 同样在血管新生内皮细胞中的表达量升高,并通过丛状蛋白PlexinD1 与Cdc42 竞争共同的效应蛋白,从而阻止VEGFR2 的降解,参与内皮细胞的定向迁移和成管[12]。
3.2 Rac1
片状伪足是细胞前缘形成的薄片状突起,富含高度动态性的分枝状微丝,是推动细胞向前运动的关键结构。Rac1 激活是驱动片状伪足凸起的主要信号,通过与质膜结合并激活WAVE,从而刺激Arp2/3 复合物,促进肌动蛋白成核和片状伪足形成[22]。胚胎Rac1 的缺失阻碍大血管的发育,并完全阻断小支毛细血管的形成,同时细胞的迁移、内腔形成、缝隙连接和通透性下降[23]。片状伪足顶端Rac1 的激活是促进细胞迁移的关键,VEGFR2介导Rac1 活化[24],活化的Rac1 通过增加内皮型一氧化氮合酶活性增加缺血组织血管新生,抑制Rac1 则减少缺血组织血管新生数量[25-26]。体外研究发现,Rac1 及其家族成员RhoG 促管腔形成的作用依赖于Cdc42 的活化,敲除Rac1 和RhoG 阻断血管生成或血管结构不稳定[27-28]。同时,Cdc42 和Rac1 均能激活P21 活化激酶(P21 activated kinase,PAK),进而激活LIM激酶(LIM kinases,LIMKs),使丝切蛋白cofilin 磷酸化从而阻断肌动蛋白解聚,提示Cdc42 和Rac1 之间存在共同信号通路。
3.3 RhoA
RhoA/Rho 相关激酶(Rho-associated protein kinase,ROCK) 信号通路为经典的G 蛋白耦联受体途径,调节细胞骨架收缩、细胞迁移和黏附。RhoA 活化成RhoA-GTP 后与ROCK 的Rho 蛋白域结合,激活ROCK,暴露ROCK 的活性中心,并向肌球蛋白轻链(myosin light chain,MLC) 转移,通过磷酸化其下游LIMKs 或MLC 影响微丝收缩,从而调节细胞骨架运动。VEGF 介导的细胞迁移依赖于RhoA/ROCK,抑制RhoA 或ROCK 均可消除VEGF 诱导的F-actin 应力纤维的形成和内皮细胞在体内外的迁移;在VEGF 刺激后,ROCK2 敲除的内皮细胞迁移和成管显著减少,这在ROCK1 敲除的内皮细胞中未被观察到,提示VEGF 诱导的内皮细胞激活主要由ROCK2 介导[29]。
在内皮细胞前沿,延伸的膜突起通过整合素黏附于细胞外基质,整合素通过RhoA/ROCK 介导的LIMKs 磷酸化,进一步激活cofilin 和转接蛋白Dia,从而稳定肌动蛋白微丝和增强肌动蛋白聚合。在内皮细胞迁移后部,ROCK 介导的肌动球蛋白收缩产生膜的拉回力,导致局部粘连的释放[30]。ROCK对细胞黏附的调节主要通过激活黏着斑激酶(focal adhesion kinase,FAK) 实现,研究发现,VEGFR2激活RhoA/ROCK 信号后,可促进FAK Tyr407 的磷酸化[31];与之不同的是,Le等[32]认为ROCK 影响FAK 的Ser732 并改变FAK 的构像,进而导致FAK 在Tyr407 上被富含脯氨酸的酪氨酸激酶2 磷酸化。
值得注意的是,RhoA/ROCK 既促血管新生,也促内皮细胞间的渗透性从而增加炎症反应。一方面,ROCK 通过抑制内皮钙黏蛋白的表达及其膜定位,促进细胞-细胞黏附连接的分离,抑制紧密连接的主要成分闭合蛋白和紧密连接蛋白1 的表达,导致内皮屏障功能障碍[33]。另一方面,ROCK 激活后可通过c-Myc 调节VEGF 转录,VEGF 本身促进内皮细胞渗透性。研究发现,ROCK 抑制剂Y-27632 以剂量依赖的方式阻断VEGF 诱导的微血管高通透性[34]。以上提示,RhoA/ROCK 在缺血性疾病中有双面性。
4 Rho GTPase 在缺血性疾病中的应用价值
血管新生为缺血性心脑血管疾病的治疗提供新方法,虽然血管新生因子促血管新生疗效令人振奋,但在基础研究向临床转化过程中其有效性和安全问题不容忽视。如Steward等[35]的两项冠心病临床试验结果均显示,尽管VEGF 组的终点事件(包括运动耐量、心绞痛分级和核素心肌灌注显像等)较安慰剂组有改善,但并未见统计差异,可能与缺血微环境中同时存在抗血管新生因子有关。此外,VEGF 在促进缺血组织血管新生的同时,加速粥样硬化斑块内的血管生成,促进斑块生长,最终可能导致斑块不稳定,出现Janus 现象[36]。生长因子作用的单一性可能是其临床效果不确定和安全问题的关键。而Rho GTPase 受GEF、GAP、GDI 等众多调节因子调控,有利于其对外部环境信号及时作出反应,可能为治疗性血管新生提供新的靶向药物。
如Cdc42 在促血管新生过程中感知信号、指导内皮细胞迁移方向和成管、稳定内皮细胞屏障。ML141 是针对Cdc42 的选择性可逆非竞争性抑制剂,ML141 与P38 激活增加有关,通过减少内皮细胞丝状伪足的长度、密度和排列,延迟眼角膜创面愈合[37]。另一种Cdc42 抑制剂ZCL367 阻碍癌细胞周期进展、增殖、迁移和肿瘤生长[38],提示Cdc42 抑制剂阻碍血管新生,但目前对Cdc42 激动剂的研究尚少,而Cdc42 激动剂可能会为缺血性疾病治疗提供方法。
RhoA/ROCK 是Rho GTPase 家族研究最详细的通路,目前临床药物主要为ROCK 的抑制剂,包括异喹啉类、砒啶类、吡唑类等,其中法舒地尔(HA1077,Y-27632) 为最早用于临床的ROCK 抑制剂。研究发现,法舒地尔既能减轻炎症反应,又通过抑制RhoA/ROCK 通路降低Drp1 蛋白表达、减少线粒体损伤从而改善心肌缺血[39]。这可能与RhoA/ROCK 下游信号通路不同有关,如Rock 通过P38 和JNK 通路增加白介素8、核因子κB(nuclear factor kappa-B,NF-κB) 等炎症 因子水平[40-41]。Y-27632 可通过抑制丝裂原活化蛋白激酶和NF-κB 减轻心肌凋亡和炎症反应,从而减小心梗大鼠梗死面积、增加血流[42],但抑制血管生成[43]。RhoA/ROCK 信号通路还促进活性氧(reactive oxygen species,ROS) 的产生,ROS 可以加速心肌梗死进程,而药物如阿托伐他汀、尼可地尔等,可通过抑制RhoA/ROCK 减轻氧化应激,保护缺血心肌细胞[44-46]。以上研究提示,RhoA/ROCK在缺血的心肌中对炎症的促进作用可能强于其对血管新生的促进作用。
5 小结
Rho GTPase 通过调节内皮细胞骨架和迁移、成管等参与血管新生过程,但同时Rho GTPase 参与炎症反应,这可能与Rho GTPase 亚家族功能结构不同、效应因子不同有关。其中,CDC42/Rac参与内皮细胞丝状伪足和片状伪足形成,促进内皮细胞向缺血缺氧组织有目的迁移和成管,但目前两者药物的研发尚处于初级阶段。RhoA/ROCK 的研究较详尽,该通路参与内皮细胞的黏附和运动,但同时也促进细胞炎症反应,ROCK 抑制剂的临床应用也主要集中在其抗炎作用。目前,在缺血性心脑血管疾病中Rho GTPase 的作用研究不多,深入了解Rho GTPase 及其调节因子、效应分子在该疾病发生发展中的功能,明确不同亚型结构在缺血组织血管新生中作用,将为缺血组织血管重建开辟新途径。
猜你喜欢
生物化工(2022年4期)2022-09-20 09:18:08
中国眼镜科技杂志(2019年9期)2019-11-11 12:15:32
安徽医科大学学报(2016年12期)2017-01-15 14:21:48
中外医疗(2015年5期)2016-01-04 03:57:57
中国病理生理杂志(2015年8期)2015-12-21 12:38:16
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
医学研究杂志(2015年11期)2015-06-10 06:44:03
恋爱婚姻家庭·养生版(2015年10期)2015-05-14 21:46:23
化工管理(2015年6期)2015-03-23 06:03:38
云南中医学院学报(2014年5期)2014-07-31 18:00:10
