
LncRNA NEAT1 调控miR-22-3p 对脂多糖诱导的人牙龈成纤维细胞生物学活性影响
2023-11-24 07:34:04范晶高一曼郑圆郭丹妮胡金龙雷小朋
医学分子生物学杂志 2023年6期
范晶 ,高一曼 ,郑圆 ,郭丹妮 ,胡金龙 ,雷小朋
1西安医学院第二附属医院口腔科 西安市,710038
2长治市第二人民医院口腔科 山西省长治市,046000
牙周炎是一种由牙菌斑堆积引发的慢性炎症性疾病,可导致牙周组织损伤,最终引起牙齿脱落[1]。该疾病不仅影响口腔健康,还与全身性疾病如心血管疾病、类风湿性关节炎、糖尿病等密切相关[2]。研究表明牙周炎的早期表现是牙龈出现炎症反应,而在牙龈结缔组织中人牙龈成纤维细胞(human gingival fibroblasts,HGFs) 最为丰富,可清除炎症细胞,其功能一旦失调,可导致炎症反应的发生,在牙周炎进展中发挥重要作用[3]。脂多糖(lipopolysaccharide,LPS) 被认为是牙周炎骨丢失的重要致病因素,虽然LPS 对牙周组织的破坏作用已被广泛认可,但具体的致病机制仍不清楚[4]。长链非编码RNA(long non-coding RNA,LncRNA) 是长度大于200 bp 的非编码RNA,参与多种生物学功能[5]。研究证明多种LncRNA 介导促炎和抗炎过程、细胞分化和存活等过程,如核富集转录本1(nuclear-enriched abundant transcript 1,Neat1) 是一种经典的LncRNA,新证据表明Neat1与炎症反应密切相关,但是否参与牙周炎的发展尚不清楚[6]。作为内源性非编码RNA 分子,微小RNA(microRNA,miRNA) 是降解mRNA 或抑制其靶基因翻译的重要介质,可作为多种疾病诊断的关键分子标志物[7]。miR-22-3p 被发现是治疗牙周炎或改善正畸引起的炎症的新靶点,但能否通过Neat1 的调控参与牙周炎的发展却鲜有报道[8]。本研究旨在探讨LncRNA Neat1 调控miR-22-3p 对LPS诱导的HGFs 生物学活性的影响。
1 材料与方法
1.1 细胞来源及主要材料、仪器
贝纳生物提供HGFs;Promega 公司提供双荧光素酶试剂盒;美国Invitrogen 公司提供LipofectmineTM2000 转染试剂、组织细胞RNA 提取试剂盒(total RNA E xtraction Kit,TRIzol);上海恪敏生物科技有限公司提供肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α) 酶联免疫吸附测定(enzymelinked immunosorbent assay,ELISA) 试剂盒;Biolegend 公司提供白细胞介素-6(interleukin-6,IL-6) ELISA 试剂盒;Sigma 公司提供LPS;Dojindo公司提供细胞增殖/毒性检测试剂盒(cell counting Kit-8,CCK-8) 试剂盒;Takara 公司提供实时荧光定量PCR(real-time fluorescence quantitative PCR,RT-qPCR) 试剂盒、逆转录试剂盒;si-NC、si-Neat1、inhibitor NC、miR-22-3p inhibitor、miR-22-3p mimics、mimics NC 及引物序列均由上海生工生物公司构建;Cell Signaling Technology 公司提供磷酸化核转录因子-κB(phosphorylated nuclear factorκB,p-NF-κB) P65 和NF-κB P65 抗体;abcam 公司提供Caspase-3 抗体;Hyclone 公司提供DMEM培养基;eBioscience 公司提供Annexin V-FITC/PI细胞凋亡试剂盒。Roche 公司提供Light Lycler 480 PCR 仪;Thermo 公司提供恒温培养箱。
1.2 细胞培养
将细胞培养在37 ℃5 % CO2的环境中,经胰蛋白酶消化后,进行传代培养。待细胞融合约90 %时,采用5 μg/mL LPS 进行诱导24 h,将细胞分为LPS 组、si-NC 组、si-Neat1 组、si-Neat1 +inhibitor NC 组、si-Neat1+miR-22-3p inhibitor 组,同时以正常培养的细胞作为对照组。si-NC 组、si-Neat1 组分别将si-NC、si-Neat1 根据LipofectamineTM2000 转染说明书转染细胞48 h 后,使用LPS进行诱导;si-Neat1+inhibitor NC 组、si-Neat1 +miR-22-3p inhibitor 组分别将si-Neat1+inhibitor NC、si-Neat1 +miR-22-3p inhibitor 共转染细胞48 h后,使用LPS 进行诱导,收集细胞进行分析。
1.3 RT-qPCR 检测Neat1、miR-22-3p 的表达水平
采用TRIzol 试剂在冰上提取总RNA,并用Nanodrop 2000 分光光度计评估RNA 样品的浓度和纯度,按照逆转录试剂盒说明书合成cDNA。根据PCR 试剂盒说明书进行实时定量RT-PCR。PCR 程序在95 ℃120 s、95 ℃20 s、60 ℃25 s 和72 ℃20 s 下进行40 个循环。miR-22-3p、Neat1 分别以U6、GAPDH 为内源性参照物,采用2-ΔΔCt计算Neat1、miR-22-3p 相对表达水平。引物序列见表1。
1.4 CCK-8 法检测细胞活力
将各组HGFs 接种于200 μL 培养基的96 孔板中(3 000 个/孔),分别培养24 h,每孔加入10 μL CCK-8 溶液,在细胞培养箱中培养2 h,在450 nm 波长处检测吸光度A值。A450nm越大表示细胞增殖活性越强。
1.5 流式细胞术检测细胞凋亡
收集细胞,用胰蛋白酶快速制备成细胞悬液,装入离心管中洗涤,然后转移到EP 管中,加入5 μL FITC 膜联蛋白Ⅴ与碘化丙啶室温避光染色10 min,使用流式细胞仪检测细胞凋亡率。
1.6 ELISA 检测炎性因子水平
收集各组细胞上清液,严格按照ELISA 试剂盒说明书测定IL-1β、TNF-α 含量。
1.7 验证Neat1、miR-22-3p 的靶向关系
先构建Neat1 的野生(Wild,WT) 和突变(Mutated,MUT) 报告质粒,然后使用LipofectamineTM2000 将细胞与构建的报告质粒与miR-22-3p mimics 或mimics NC 共转染,48 h 后测量荧光素酶活性。
1.8 蛋白质印迹检测相关蛋白表达水平
使用RIPA 裂解缓冲液提取各组蛋白质并检测其浓度,SDS-PAGE 分离蛋白质并转移到膜上,将其与5 %脱脂牛奶一起孵育1 h,并在4 ℃下与caspase-3、p-NF-κB P65、NF-κB P65 一抗孵育过夜。次日,将膜置于二抗中室温孵育1 h。ECL 可视化蛋白,以β-actin 为内参,分析各组蛋白表达水平。
1.9 统计学处理
2 结果
2.1 比较各组间Neat1、miR-22-3p 表达水平
与对照组相比较,LPS 组Neat1 表达显著增加,miR-22-3p 表达显著降低(P<0.05)。
与LPS 组、si-NC 组相比较,si-Neat1 组Neat1的表达显著性降低,miR-22-3p 的表达显著性增加(P<0.05)。
与si-Neat1 +inhibitor NC 组相比较,si-Neat1 +miR-22-3p inhibitor 组Neat1 的表达显著性增加,miR-22-3p 的表达显著性降低(P<0.05)(表2)。
表2 比较各组细胞中Neat1、miR-22-3p 表达水平(, n=6)
与对照组比较,aP<0.05,与LPS 组比较,bP<0.05;与si-NC 组比较,cP<0.05;与si-Neat1 +inhibitor NC 组比较,dP<0.05
2.2 比较各组间的细胞增殖活性
与对照组比较,LPS组A450nm显著降低(P<0.05);与LPS 组、si-NC 组比较,si-Neat1组A450nm显著增加(P<0.05);与si-Neat1+inhibitor NC 组比较,si-Neat1 +miR-22-3p inhibitor组A450nm显著降低(P<0.05)(表3)。
表3 比较各组间细胞的增殖活性(, n=6)

表3 比较各组间细胞的增殖活性(, n=6)
与对照组比较,a P<0.05,与LPS 组比较,b P<0.05;与si-NC 组比较,cP<0.05;与si-Neat1 +inhibitor NC 组比较,dP<0.05
2.3 比较各组间的细胞凋亡情况
与对照组比较,LPS 组细胞凋亡率显著增加(P<0.05);与LPS 组、si-NC 组比较,si-Neat1 组细胞凋亡率显著下降(P<0.05);与si-Neat1 +inhibitor NC 组比较,si-Neat1 +miR-22-3p inhibitor 组细胞凋亡率显著增加(P<0.05)(图1,表4)。
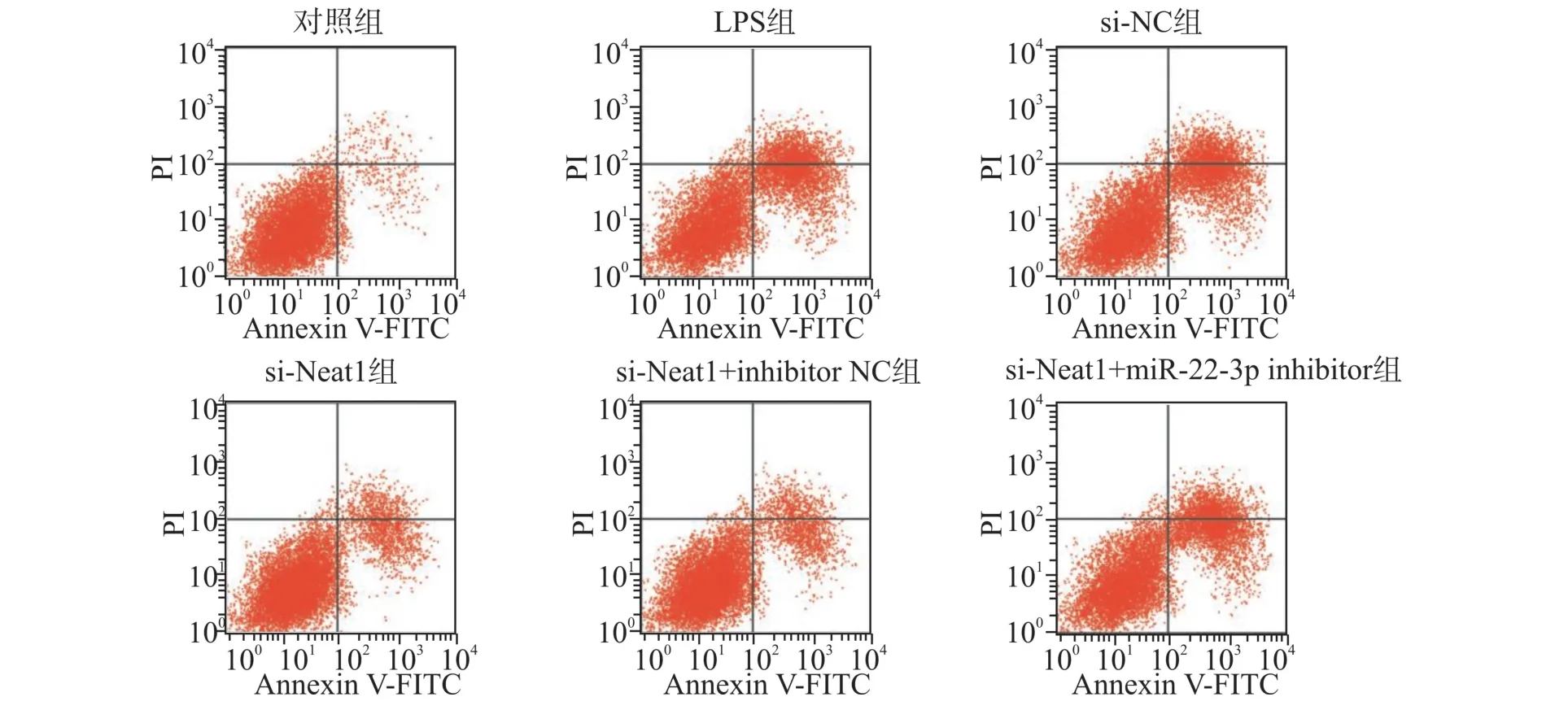
图1 各组细胞凋亡变化
表4 比较各组间细胞凋亡率(, n=6)
与对照组比较,aP<0.05,与LPS 组比较,bP<0.05;与si-NC 组比较,cP<0.05;与si-Neat1+inhibitor NC 组比较,dP<0.05
2.4 比较各组间的炎症因子水平
与对照组比较,LPS 组IL-1β、TNF-α 水平显著增加(P<0.05)。
与LPS 组、si-NC 组比较,si-Neat1 组IL-1β、TNF-α 水平显著下降(P<0.05)。
与si-Neat1+inhibitor NC 组比较,si-Neat1 +miR-22-3p inhibitor 组IL-1β、TNF-α 水平显著增加(P<0.05)(表5)。
表5 比较各组间IL-1β、TNF-α 水平(, n=6)

表5 比较各组间IL-1β、TNF-α 水平(, n=6)
与对照组比较,aP<0.05,与LPS 组比较,bP<0.05;与si-NC 组比较,cP<0.05;与si-Neat1 +inhibitor NC 组比较,dP<0.05
2.5 验证Neat1、miR-22-3p 的靶向关系
TargetScan 显示Neat1、miR-22-3p 存在结合位点(图2)。与mimics NC+Neat1 WT 组比较,miR-22-3p mimics+Neat1 WT 组荧光素酶相对活性降低(P<0.05)(表6)。
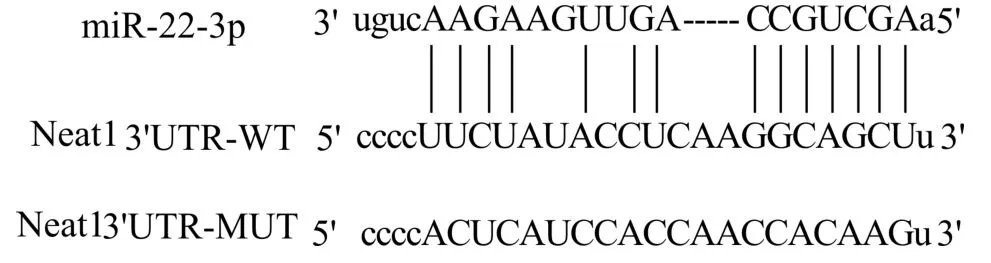
图2 TargetScan 数据库预测Neat1、miR-22-3p 的结合位点
表6 靶向验证Neat1、miR-22-3p 的关系(, n=6)

表6 靶向验证Neat1、miR-22-3p 的关系(, n=6)
与mimics NC+Neat1 WT 组比较,∗P<0.05
2.6 比较各组细胞相关蛋白表达
与对照组比较,LPS 组Caspase-3、p-NF-κB P65/NF-κB P65 表达增加(P<0.05);与LPS 组、si-NC 组比较,si-Neat1 组Caspase-3、p-NF-κB P65/NF-κB P65 表达下降(P<0.05);与si-Neat1+inhibitor NC 组比较,si-Neat1+miR-22-3p inhibitor 组Caspase-3、p-NF-κB P65/NF-κB P65 表达增加(P<0.05)(图3、表7)。
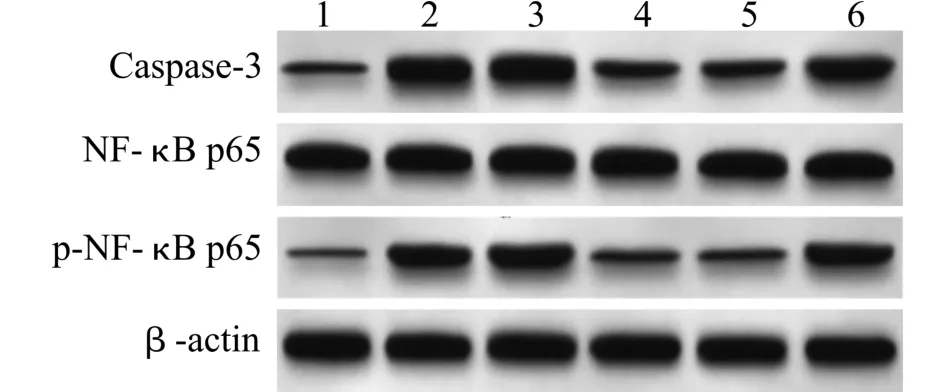
图3 蛋白质印迹检测各组细胞p-NF-κB P65、NF-κB P65、Caspase-3 蛋白表达
表7 比较各组间p-NF-κB P65/NF-κB P65、Caspase-3 蛋白表达(,n=6)
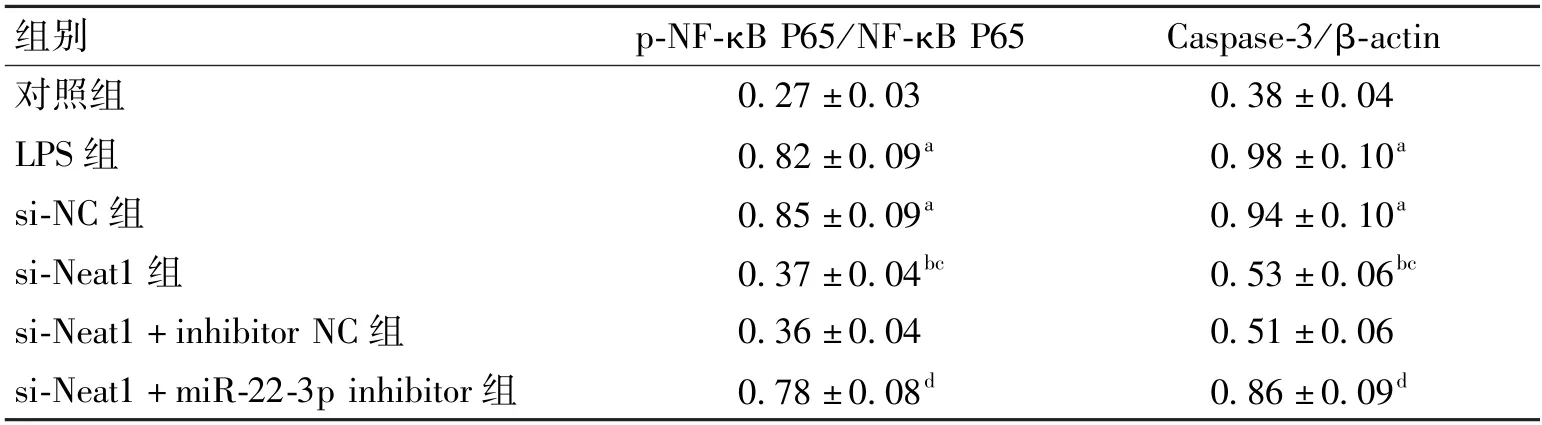
表7 比较各组间p-NF-κB P65/NF-κB P65、Caspase-3 蛋白表达(,n=6)
与对照组比较,aP<0.05,与LPS 组比较,bP<0.05;与si-NC 组比较,cP<0.05;与si-Neat1 +inhibitor NC 组比较,dP<0.05
3 讨论
牙周炎是一种严重的口腔疾病,以感染引起的牙龈组织炎症为特征,逐渐导致骨骼支撑、结缔组织的丧失,最终使人牙齿脱落[9]。牙龈卟啉单胞菌是引起慢性牙周炎发生发展的牙周致病菌,而LPS 是该病菌细胞壁的主要组成成分,可激发宿主细胞免疫反应,作为一种重要的毒力因子,破坏牙周组织,加重牙周炎进程[10]。本实验以LPS 诱导HGFs 建立牙周炎损伤,结果发现LPS 诱导HGFs,促进了炎症反应和细胞凋亡,严重影响了细胞生长,可进一步对其作用机制进行探索。
长链非编码RNA 是长度超过200 个核苷酸的非编码RNA,参与炎症性疾病、肿瘤和呼吸系统疾病等多种疾病的发生[11]。Neat1 作为一种LncRNA,参与了肿瘤发生、免疫反应和感染等许多病理生理过程[12]。最新研究表明Neat1 在炎症免疫调节中也发挥关键作用。如特应性皮炎小鼠模型中Neat1 表达显著增加[13],沉默NEAT1 可明显改善特应性皮炎小鼠症状,抑制通路相关因子的释放。黄舒颖等[14]研究报道了在LPS 诱导的NR8383 肺泡巨噬细胞中,沉默LncRNA Neat1 表达可以显著降低炎症因子水平,为脓毒症引起的肺部损伤提供治疗方案。Neat1 在炎症性肠病小鼠模型中表达上调[15],并通过调节外泌体介导的巨噬细胞调节肠道炎症,敲除Neat1 可以缓解小鼠肠道炎症。本研究发现LPS 诱导HGFs 中,Neat1 表达上调,沉默Neat1表达可明显抑制炎症反应及细胞凋亡,提高细胞活力,表明沉默Neat1 表达有助于缓解LPS 诱导HGFs的炎症作用,促进细胞生长。
LncRNAs 可作为ceRNAs 调节靶向mRNA 的表达。TargetScan 数据库显示Neat1 与miR-22-3p 存在结合位点,且双荧光素酶报告基因验证了两者的靶向关系。miR-22-3p 表达异常与炎症性疾病相关。如miR-22-3p 在脓毒症诱导的急性肾损伤大鼠模型和LPS 诱导的HK-2 细胞中均显着下调[7],miR-22-3p 过表达通过靶向PTEN 在体内和体外抑制炎症反应和细胞凋亡。在LPS 诱导的HK-2 细胞炎症模型中[16],miR-22-3p 模拟物显著降低炎症细胞的自噬和凋亡,为急性肾损伤的研究提供方向。本研究发现LPS 诱导HGFs 中,miR-22-3p 表达水平下降,与Zheng等[8]研究结果吻合,表明可能与牙周炎的发生有关。转染si-Neat1 后,miR-22-3p 表达水平升高,两者出现负靶向调控,缓解炎症反应;当共转染si-Neat1 +miR-22-3p inhibitor 后,miR-22-3p 表达水平降低,Neat1 表达升高,炎症反应加重,提示沉默Neat1 表达可通过上调miR-22-3p 表达减轻炎症反应、细胞凋亡,实现对HGFs 的保护。另外,对于炎症疾病、应激反应以及免疫应答等过程,NF-κB信号通路极其重要,包括在牙周炎中NF-κB 可刺激IL-1β、TNF-α 等炎症因子表达,导致组织受损[17]。张娜等[18]研究表明在LPS 刺激的HGFs 研究中,甘草甜素发挥抗炎作用,与抑制NF-κB 通路表达密切相关。本研究发现沉默Neat1 表达可通过上调miR-22-3p 表达进而抑制NF-κB 表达,降低炎症反应,缓解疾病。
综上所述,沉默Neat1 表达可抑制LPS 诱导HGFs 的炎症反应、细胞凋亡,进而保护HGFs,其机制可能通过上调miR-22-3p 表达,抑制NF-κB 表达有关,为牙周炎治疗提供实验依据,但由于miR-22-3p 下游靶点较多,机制较为复制,相关研究还在进行中。
猜你喜欢
保健医苑(2022年5期)2022-06-10 07:46:38
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
昆明医科大学学报(2021年6期)2021-07-31 07:40:38
肝博士(2020年5期)2021-01-18 02:50:18
实用口腔医学杂志(2017年6期)2017-09-19 02:51:32
中国继续医学教育(2015年4期)2016-01-07 07:38:01
法医学杂志(2015年4期)2016-01-06 12:36:36
法医学杂志(2015年4期)2016-01-06 12:36:36
中国医疗美容(2015年1期)2015-07-12 10:06:47
医学研究杂志(2015年7期)2015-06-22 11:01:01
