
WGCNA与机器学习算法识别结直肠癌发病机制中潜在PANoptosis关键基因
2023-11-18 10:17李文静杨杰陈冬梅
东南大学学报(医学版) 2023年5期
李文静,杨杰,陈冬梅,2
(1.贵州医科大学,贵州 贵阳 550000; 2.六盘水市人民医院 消化内科,贵州 六盘水 553000)
结直肠癌(CRC)作为一种常见的消化道恶性肿瘤,其发病率和死亡率位居肿瘤第3位和第2位[1]。尽管CRC的5年生存率已有明显提高,但目前中国CRC的发病率和死亡率仍呈上升趋势且有年轻化倾向[2]。由于CRC的早期症状无特异性,易被误诊为痔出血、息肉出血等,部分患者在确诊时已处于中晚期。因此,早期诊断是改善CRC患者预后的关键。结肠镜检查是诊断CRC的最典型方法,具有较高特异性和敏感性,但为侵入性检查,具有价格高、条件苛刻等缺点,患者接受性差[3]。以癌胚抗原(CEA)为代表的非侵入性生物标志物诊断CRC的特异性和敏感性不理想[4]。筛选高敏感性和特异性的生物标志物对于CRC的筛查、诊疗和预后预测具有重要意义。
PANoptosis是近来定义的一个新概念,强调细胞凋亡、焦亡和坏死性凋亡之间的串扰和协调[5]。既往针对CRC与各种形式细胞死亡相关的基因已经在许多研究中被报道,包括自噬、焦亡、坏死性凋亡相关基因。然而,与不同类型细胞死亡之间串扰和协调相关基因的研究仍不充分。生物信息学是结合了生物学、数学和信息技术的新兴学科。利用生物信息学方法筛选肿瘤相关分子生物标志物在潜在的高危患者诊断、鉴定和预后评估中显示出卓越的性能[6]。加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)作为生物信息学分析中被高频应用的一种方法,在筛选疾病特征生物标志物和潜在防治靶点中表现出明显优势,在包括肿瘤在内的多种疾病中应用广泛[7]。机器学习算法作为人工智能的子集之一,被广泛应用于疾病生物标志物、发病机制、治疗靶点等方面的研究[8]。本研究基于高通量测序数据,利用WGCNA与机器学习算法相结合的方法,筛选与CRC发病相关的潜在PANoptosis关键基因并构建疾病预测模型,以期为CRC的诊断和治疗提供一定指导依据。
1 资料与方法
1.1 数据集的下载及数据处理
从美国国立生物技术信息中心(NCBI)的GEO芯片数据库中选择了GSE39582和GSE74602 2个芯片数据集,从中获取CRC和正常黏膜组织的基因表达谱。GSE39582采用美国昂菲公司人基因组U133 Plus 2.0芯片GPL570平台,包括443个肿瘤样本和19个对照样本。GSE74602采用美国昂菲公司人基因组Ref8 2.0芯片GPL6104平台,包括30个肿瘤样本和30个对照样本。下载芯片数据压缩包和探针文件,通过R4.2.3软件的RMA算法对芯片数据进行标准化处理。采用|logFC|>1和adj.p<0.05作为筛选标准,使用R软件的limma包进行差异表达基因(DEGs)的筛选。
1.2 功能富集分析
分别采用R软件enrichDO函数、clusterProfiler包对DEGs进行疾病本体(DO)、基因本体(GO)和基因组京都百科全书(KEGG)富集分析。其中GO分析包括了差异基因的细胞组成(CC)、分子功能(MF)和生物学过程(BP)。结果根据-log10(P)值大小进行排序,同时呈现富集基因占总体的比例。
1.3 WGCNA
采用R软件WGCNA包进行加权基因共表达网络识别hub基因。首先,计算所有基因的表达相关系数,构建相似矩阵。将相似矩阵转化为拓扑矩阵,采用拓扑重叠度量(TOM)描述基因之间的关联度。根据基因间相关性确定软阈值(power),构建基因聚类树。根据基因表达相关系数确定表达模块,每个模块内的最少基因数设定为100个,模块间相关系数设定为0.85。特征基因由模块内基因连通性决定。根据基因表达趋势计算皮尔逊相关系数,取相关系数最大的模块内基因作为特征基因。
1.4 机器学习算法
利用R软件的venn包取DEGs和WGCNA特征基因的交集基因。利用R软件glmnet包的最小绝对值收敛和选择算子(LASSO)算法、e1071包的支持向量机-递归特征消除(SVM-RFE)、RandomForest包的随机森林算法从交集基因中获得枢纽基因。
1.5 Panoptosis关键基因的筛选
基于geneCards(https:∥www.genecards.org/),分别以“Pyroptosis”“Necroptosis”和“Apoptosis”为关键词获得焦亡、坏死和凋亡相关基因。利用R软件的venn包取焦亡、坏死和凋亡相关基因的交集,获得PANoptosis相关基因(PANRGs)。利用R软件的venn包,取PANoptosis相关基因和枢纽基因的交集,获得PANoptosis关键基因。
1.6 诊断列线图模型的构建和验证
以GSE39582作为训练集,GSE74602为验证集,基于PANoptosis关键基因在训练集和验证集中的表达量,使用R软件rms包构建预测CRC的列线图模型。使用校准曲线评估列线图模型的预测能力。采用决策曲线评估列线图模型的临床效用。使用R包pROC函数创建受试者工作特征(ROC)曲线,以确定PANoptosis关键基因和列线图模型在训练集和验证集中对CRC的诊断价值。
2 结 果
2.1 DEGs筛选结果与功能富集分析
根据adj.p<0.05和|log2FC|>1筛选标准,从GSE39582中筛选出1 503个DEGs,其中上调744个,下调759个,见图1A。DO分析(图1B)显示,DEGs主要与良性肿瘤、肾细胞癌、乳腺癌、肠道疾病等多种肿瘤有关。GO分析结果(图1C)表明,DEGs主要参与核分裂、染色体分离、有丝分裂的核分裂等生物学过程;主要定位于含胶原蛋白的细胞外基质、细胞的顶端部分、顶端质膜等细胞成分;主要参与细胞外基质结构成分、糖胺聚糖结合、细胞因子活性、CXCR趋化因子受体结合等分子功能。KEGG分析(图1D)显示,DEGs主要富集于细胞周期、嘌呤代谢、TNF信号通路、PPAR信号通路、P53信号通路等相关信号通路。
2.2 加权基因共表达网络构建
使用R软件“WGCNA”包对GSE39582中的样本进行聚类(图2A)。随后进行无标尺度指数和平均连通性分析,当软阈值β=4时,网络达到无标尺度拓扑阈值0.9,且网络结点平均连接性高(图2B)。通过动态树切割和计算,获得8个基因模块。将8个模块与数据集对照组和肿瘤组之间进行相关性分析,得到相关性热图(图2C),选择与肿瘤相关性最强的棕色模块(r=-0.65,P<0.001)为CRC的关键模块。根据基因显著性的过滤条件为0.5,以及基因与模块相关性的过滤条件为0.8,确定棕色模块中核心基因820个(图2D)。
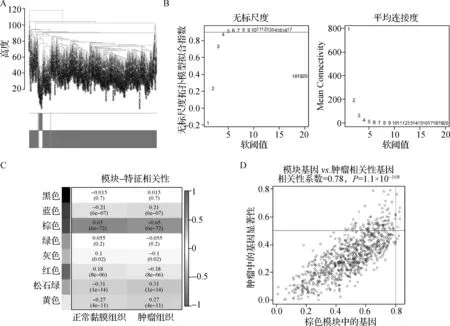
A.样本-性状聚类树;B.软阈值参数与尺度独立性、平均连接性分析(最佳软阈值为4);C.临床性状与基因模块的相关性热图,单元格中的数字分别表示相关性系数r和对应的P值;D.棕色模块基因散点图
2.3 特征差异基因的确定及功能富集分析
根据基因差异表达分析和WGCNA的结合,共有1 503个DEGs和820个关键模块基因,二者取交集,得到525个交集基因(图3)。对这525个交集基因进行富集分析,DO富集分析共富集到422条DO条目,涉及肠道疾病、胃癌、结肠炎、炎症性肠病。GO分析共富集到1 637条GO条目,这些交集基因在醇类代谢过程、四氯化碳运输、细胞顶端部分、顶端质膜、阴离子跨膜转运体活性、次级活性跨膜转运体活性等方面显著富集。KEGG分析表明,这些交集基因涉及紧密连接、视黄醇代谢、药物代谢等途径。
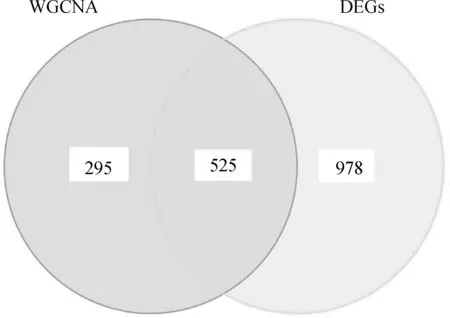
图3 特征差异基因的确定及功能富集分析
2.4 基于3种不同机器学习算法筛选枢纽基因
在GSE39582中,利用SVM-RFE对上述525个特征差异基因进行筛选,“最佳点”在0.003 39时5折交叉验证误差最小,共筛选出8个特征基因(图4A)。随机森林算法获得30个特征基因(图4B)。LASSO回归分析显示,在选择最小均方误差对数λ时,回归模型中有9个系数不为零的特征基因(图4C)。将SVM-RFE、RandomForest和LASSO分析所筛选的基因取交集,共得到6个枢纽基因(HubGenes,图4D),即细胞周期蛋白依赖性激酶1(CDK1)、二肽酶1(DPEP1)、水通道蛋白8(AQP8)、溶质载体家族30成员10(SLC30A10)、半胱氨酸天冬氨酸蛋白水解酶 7(CASP7)和半胱氨酸天冬氨酸蛋白水解酶 8(CASP8)。
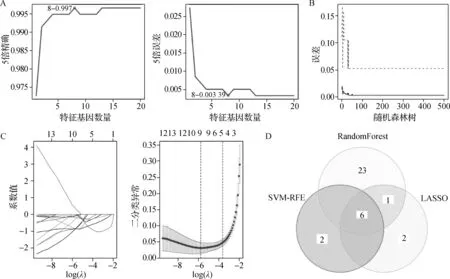
A.SVM-RFE分析;B.Random Forest分析;C.LASSO回归分析;D.SVM-RFE、Random Forest和LASSO交叉分析的韦恩图
2.5 PANoptosis关键基因的筛选与功能富集分析
以“Pyroptosis”为关键词,在geneCards中获得焦亡相关基因436个。以“Necroptosis”为关键词,在geneCards中获得坏死相关基因667个。以“Apoptosis”为关键词,在geneCards中获得凋亡相关基因14 989个。取交集,得到PANoptosis相关基因(PANRGs)96个(图5A)。进一步取PANRGs和HubGenes的交集,获得4个PANoptosis关键基因,即CDK1、DPEP1、CASP7和CASP8(图5B)。
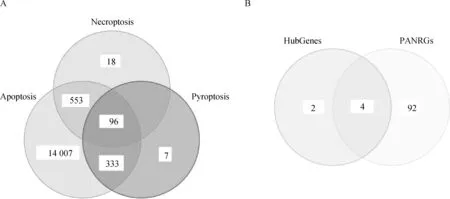
A.焦亡、坏死和凋亡相关基因的交集,获得PANoptosis相关基因(PANRGs);B.取PANRGs和枢纽基因(HubGenes)的交集,获得PANoptosis关键基因
2.6 CRC预测模型构建与验证
基于4个PANoptosis关键基因,在训练集GSE39582中采用Logistic回归分析构建nomogram图(图6A)。校准图显示,校准预测曲线与标准曲线贴合较好。决策(DCA)曲线显示,列线图模型在预测CRC发生的临床效能上表现良好。在验证集GSE74602中构建的列线图、校准图和DCA曲线也证实了上述结果(图6B)。

A.训练集GSE39582中基于PANoptosis关键基因构建的列线图模型、校准曲线和DCA曲线;B.验证集GSE74602中基于PANoptosis关键基因构建的列线图模型、校准曲线和DCA曲线
2.7 PANoptosis关键基因和预测模型的诊断效能评价
在训练集GSE39582中,CDK1、DPEP1、CASP7和CASP8的AUC分别为0.926、0.953、0.991和0.873,而预测模型的AUC为0.998。在验证集GSE74602中,CDK1、DPEP1、CASP7和CASP8的AUC分别为0.999、0.931、0.941和0.651,而预测模型的AUC为1.000。说明预测模型能够准确地区分正常和肿瘤组织样本。见图7。
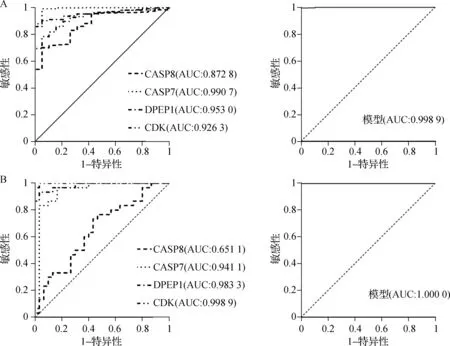
A.在训练集GSE39582中CDK1、DPEP1、CASP7、CASP8和列线图模型区分肿瘤组织和正常组织的ROC曲线;B.在验证集GSE74602中CDK1、DPEP1、CASP7、CASP8和列线图模型区分肿瘤组织和正常组织的ROC曲线
3 讨 论
CRC早期诊断困难,治疗效果及预后均不理想,且详细的分子生物学机制尚未完全阐明。探究新的分子标志物和分子治疗靶点对研究CRC的发病机制、预防和治疗具有重要意义。近年来,微阵列和生物信息学分析已被广泛用于鉴定不同癌症诊断和治疗的潜在靶标[9]。本研究基于差异基因表达筛选与WGCNA共鉴定出525个疾病特征DEGs,主要与涉及肠道疾病、胃癌、结肠炎、炎症性肠病等疾病。此外,它们可以通过调控醇类代谢过程、四氯化碳运输、药物代谢等生物学功能和途径介导CRC的病理过程。这些结果证实了这些重要DEGs介导CRC的分子机制与既往文献报道结果存在一些吻合[10],在一定程度上证实了本研究结果的准确性。
机器学习算法能够从新视角来分析大群体基因测序或微阵列数据。随机森林、LASSO和SVM等机器学习算法解决了既往高维数据构建的预测模型可能具有的过度拟合风险等问题[11]。与使用组学生物标志物组合的数学方法相比,机器学习算法定义风险等级在疾病预测中表现出更优的性能[12]。因此,集合多种机器学习算法开发与CRC病理机制相关的生物标志物来预测CRC有重要临床意义。本研究利用LASSO、SVM-RFE和RandomForest算法从525个特征差异基因中鉴定出了6个CRC的枢纽基因,即CDK1、DPEP1、AQP8、SLC30A10、CASP7和CASP8。
肿瘤的发生发展是一个与多基因相关的多步骤、多阶段复杂过程。目前研究认为,肿瘤是细胞增殖和死亡失衡所致。其中,细胞死亡在肿瘤发生发展过程中起负调控作用,可以遏制肿瘤细胞的快速生长。在程序性细胞死亡的拟议形式中,细胞凋亡、焦亡和坏死性凋亡已被广泛研究,这些过程涉及负责细胞死亡的启动、转导和执行的复杂分子机制[13]。然而,关于细胞死亡的早期研究通常集中于这些独立机制下的独特程序和功能。PANoptosis是最近发现的一种程序性细胞死亡类型,可促进肿瘤细胞死亡,并可能降低异常凋亡途径对肿瘤化学抗性的影响[14]。作为一种依赖于PANoptosome复合物的炎症程序性细胞死亡,PANoptosis含有焦亡、细胞凋亡和坏死性凋亡所必需的分子,可激活不同的细胞死亡途径[15]。即使其他程序性细胞死亡途径被抑制或突变,PANoptosis也可以提供另一种途径来杀死癌细胞并降低癌细胞获得耐药性的风险[16]。因此,探讨关于PANoptosis对癌症影响的机制对于开发癌症治疗新策略至关重要。Huang等[17]筛选了与CRC预后特征相关的PANoptosis相关基因,并提出基于PANoptosis相关基因构建的风险模型有望成为CRC的潜在预后标志物。本研究进一步取枢纽基因与PANoptosis相关基因的交集,得到4个与CRC相关的PANoptosis关键基因,即CDK1、DPEP1、CASP7和CASP8。CDK1是丝氨酸/苏氨酸蛋白家族成员之一,已被证实参与细胞周期的调控。研究证明,CDK1可作为预测Ⅱ期CRC复发的指标,且其高表达与较高的远处转移率和较低的病因特异性生存率显著相关[18]。DPEP1是由肽键连接的两个氨基酸残基组成的一种二肽化合物,可在胰腺、乳腺、结直肠等多种组织中表达[19]。研究[20]显示,DPEP1通过调节基因转录调控通路来发挥致癌作用。黄金华等[21]认为,DPEP1在CRC不同病理阶段的表达有助于CRC的早期诊断。CASP7是凋亡通路关键基因,在控制细胞死亡中充当环境传感器,其遗传变异已被证实与CRC患者放化疗敏感性和副反应及生存密切相关[22]。CASP8诱导了广泛的生物学过程,已被证实是一种控制焦亡、细胞凋亡和坏死性凋亡的分子开关[23]。
与单独使用一种机器学习算法相比,整合多种机器学习算法构建的疾病预测标志物具有更优异的性能[24]。Liu等[25]发现,整合LASSO和SVM算法构建的预后标志物组合表现出对乳腺癌生存率预测更强的效能。本研究基于上述4个与CRC相关的PANoptosis关键基因构建了列线图模型,发现该列线图模型在预测CRC发生的临床效能上表现良好。进一步采用训练集和验证集评估了列线图模型诊断CRC的ROC曲线,发现AUC值均大于0.9,且其预测效能优于各单基因,说明基于这4个PANoptosis关键基因的预测模型在CRC诊断方面具备潜在的优势。然而,本研究仍存在几个局限性。首先,本研究仅纳入了GEO数据库中2个数据集样本,未与其他数据库进行联合比较,无法排除转录本可能存在的选择偏倚。其次,未对4个PANoptosis关键基因在CRC细胞中的功能和潜在机制进行评估,仍需要额外的体内和体外实验来进行功能和临床验证。此外,在临床样本中对所筛选的基因和构建的模型进行验证是十分必要的。
综上所述,通过综合运用WGCNA和LASSO、SVM-RFE和RandomForest算法鉴定了4个PANoptosis关键基因,开发并验证了基于这4个基因的列线图预测模型。结果显示,构建的列线图模型可能成为诊断CRC的有价值工具。本研究为后续进一步深入探讨CRC的发病机制,开发新型诊断生物标志物提供了新思路。
猜你喜欢
现代临床医学(2023年4期)2023-09-26
中学生数理化(高中版.高考理化)(2021年6期)2021-07-28
供水技术(2020年6期)2020-03-17
东北史地(学问)(2016年6期)2016-12-14
中华老年多器官疾病杂志(2016年9期)2016-04-28
医学研究杂志(2015年7期)2015-06-22
现代检验医学杂志(2015年6期)2015-02-06
现代检验医学杂志(2015年5期)2015-02-06
NBA特刊(2014年7期)2014-04-29
中国商人(2013年1期)2013-12-04
