
高效电解二氧化锰电化学分析研究
2023-11-17 07:02裴启飞卢文鹏郭孟伟邵伟春王恩泽张启波
有色金属科学与工程 2023年5期
裴启飞, 卢文鹏, 郭孟伟, 邵伟春, 王恩泽, 张启波*
(1.云南驰宏锌锗股份有限公司,云南 曲靖 655011; 2.昆明理工大学冶金与能源工程学院, 昆明 650093)
二氧化锰(MnO2)具有多种晶体结构和良好的电化学活性等[1-3],在电催化/光电催化、污染物降解、储能材料等领域有着广泛应用[4-7]。根据制备方法,可将MnO2分为天然二氧化锰(Natural Manganese Dioxide, NMD)[8]、电解二氧化锰(Electrolytic Manganese Dioxide, EMD)[9]和化学二氧化锰(Chemical Manganese Dioxide, CMD)[10]。不同制备方法所获得的MnO2在晶体结构、结晶水含量、微观形貌和纯度等方面均存在显著差异[11-13];与其他制备方法相比,电化学沉积是批量化制备高纯MnO2的有效方法[14-16]。
基于不同的电解体系,EMD的制备方法主要有硫酸法、氯化法、硝酸法、碳酸法和“两矿法”等[17-19]。在众多电解体系中,硫酸体系具有廉价、低腐蚀性和无毒性气体排放等优势,是制备EMD的主要介质[20]。然而,锰(Mn)作为典型的变价金属,存在多种氧化形态(MnO、Mn2O3、Mn3O4、MnO2等),为实现MnO2的电化学高效、定向制备,需严格控制工艺条件。电化学研究表明[21],EMD的电解过程涉及[Mn(H2O)6]ads3+中间体的形成,并以水解的方式生成MnOOH沉淀,进一步通过固态氧化转变成MnO2。事实上,MnO2的电解过程还存在析氧、生成混合锰氧化物和MnO4-等多种潜在副反应,导致EMD的电流效率偏低,产品品质不稳定。已有研究表明,EMD的高效制备受温度、电极材料、电流密度、硫酸浓度和Mn2+浓度、杂质离子含量等诸多因素影响[14,19,22],但多数报道主要围绕工艺优化开展研究,缺乏对相关的电化学理论分析。本研究基于电化学分析方法,系统地探究了硫酸体系电解MnO2过程中Mn2+的电化学行为,揭示体系pH、电位、温度等对电极反应过程的影响规律,为进一步优化电解体系和沉积参数提供理论基础,指导EMD长周期、稳定高效电解。
1 实验部分
1.1 实验试剂和材料
MnSO4·H2O(纯度>99%,上海阿拉丁生化科技股份有限公司),H2SO4(分析纯,重庆川东化工(集团)有限公司)。采用去离子水作为溶剂,将不同浓度的H2SO4和MnSO4·H2O经混合溶解后,配制成所需电解液。工作电极和对电极均采用高纯石墨片(厚度为0.5 cm),以Hg/Hg2SO4电极(填充饱和K2SO4溶液,E(RHE) = 0.652 +E(vs. Hg/Hg2SO4) + 0.059 ×pH)为参比电极。电解实验中,采用蠕动泵(LongerPump,BT300-1F,YZ1515x)实现电解液的循环,以商用氟胶管(圣戈班,A-60-G17#)连接循环系统,体系循环速率为300 mL/min。
1.2 电化学测试
电化学测试方法主要包括线性伏安法、循环伏安法和交流阻抗法等,所有电化学测试均在CHI 760E型电化学工作站(上海辰华公司)上进行。
采用线性伏安法(Linear Sweep Voltammetry,LSV)探索Mn2+的电化学氧化行为,明确Mn2+→MnO2的热力学条件,确定合适的电位区间。采用三电极体系进行LSV测试,以双石墨棒(ϕ= 0.5 cm)分别做工作电极和对电极,Hg/Hg2SO4电极为参比电极,测试电位范围为0~1.4 Vvs. Hg/Hg2SO4,扫描速度为5 mV/s。类似地,采用双电极体系进行LSV测试时,以双石墨棒(ϕ= 0.5 cm)分别做阴极和阳极,电压范围设置为1~3 V。
采用循环伏安法(Cyclic Voltammetry,CV)探究Mn2+的氧化还原行为。选用三电极体系进行CV测试,以双石墨棒(ϕ= 0.5 cm)分别做工作电极和对电极,Hg/Hg2SO4电极作为参比电极,扫描速度为5 mV/s。
基于电化学交流阻抗谱(Electrochemical Impedance Spectroscopy,EIS)测定Mn2+→ MnO2转化过程中的界面电荷转移情况。EIS测试所施加的电位值设定为0.8 Vvs. Hg/Hg2SO4,扫描频率范围为100 kHz~0.05 Hz,交流振幅扰动为5 mV。
1.3 紫外可见光谱测试
采用Varian Cary 50紫外可见分光光度计对电解实验前后的电解液进行紫外可见光谱(UV-vis spectrum)测试,对比分析施加电流密度对MnO2电解过程的影响。UV-vis测试在径长为1 cm的比色皿中进行,波长范围为200~800 nm,扫描速率为60 nm/min。
1.4 电解实验
H2SO4-MnSO4体系中电解MnO2所用阴极和阳极均为高纯石墨板(长5 cm,宽5 cm,厚0.5 cm,面积60 cm2)。将Hg/Hg2SO4参比电极与阳极并联,引入数显万用表(B35T)对阳极电位进行监测。采用直流电源(UTP1306S)供电,用数显恒温水浴锅对溶液进行加热和保温。使用定制电解槽进行电解实验,其尺寸为30 cm(长)×15 cm(宽)×10 cm(高),并外接3 L高位槽结合蠕动泵对电解液进行循环。电解实验采用的电解装置以平行排布的阴极和阳极石墨板搭建。MnO2的电解效率(η(MnO2))计算式如下所示:
其中:Δm1和Δm2分别为电解MnO2计算的理论增重和获得的实际增重;i为电解时施加的阳极电流;t为电解时间;n为Mn2+→ MnO2氧化过程所转移的电子数(2);F为法拉第常数(96 485 C/mol);M(MnO2)为MnO2的相对分子质量(87 g/mol)。
1.5 电解产物表征
采用粉末X-射线衍射仪(X-ray diffraction,XRD)对电解产物的晶体结构进行分析,探索不同条件对产物结晶度的影响。
2 结果与讨论
2.1 热力学分析
图1所示为25 ℃下Mn-H2O体系的E-pH图[23]。由图1可知,锰氧化物生成的物种依赖于体系的pH和阳极电位。为定向生成MnO2,必须将电解条件控制在图1中灰色区域(pH<3),从而避免生成混合锰氧化物;特别地,当溶液pH<0(H2SO4含量>50 g/L)时,MnO2的优势生成区处于H2O的热力学稳定区域之上。理论上,生成MnO2的同时会导致析氧。然而,在酸性条件下,阳极析氧反应存在很高的过电位,使得O2的实际析出电位远大于理论值(通常超出1.0 V),从而为MnO2的高效电解提供了可能。

图1 25 ℃下Mn-H2O体系的E-pH图[23]Fig.1 E-pH diagram of Mn-H2O system at 25 ℃[23]
MnO2电解过程涉及的电化学反应如式(1)—式(4)所示。根据电荷平衡原理,每消耗2 mol电子可生成1 mol MnO2,在阳极处产生4 mol H(+式(2));在相同电荷下,阴极处仅能消耗2 mol H+(式(1))。对于整个反应过程,阴/阳极上的氧化还原过程将额外生成2 mol H+,长时间电解将会导致溶液pH下降。
由图1可知,溶液pH下降对MnO2的生成不利。pH越小,生成MnO2的阳极电位越高,所需能耗越大。此外,阳极电位过高会发生副反应,生成MnO-4(式(3))并析氧(式(4)),从而大大降低电解MnO2的效率。因此,为实现MnO2的高效电解,需严格控制体系pH和阳极电位,以避免副反应的发生。
阴极反应[15,24]:
阳极主反应:
阳极副反应:
为了探索合适的溶液pH范围,探索了H2SO4浓度对阴极析氢与阳极Mn2+→ MnO2氧化反应的影响。图2显示了10、50、100、150 g/L H2SO4溶液中含12.55 g/L Mn2+的动电位极化曲线(LSV)。在40 ℃下,随着H2SO4浓度逐渐升高,Mn2+的氧化峰逐渐正移,表明Mn2+的氧化在热力学上变得困难, 这与E-pH图分析结果一致。同时,对于阴极析氢反应,随着H2SO4浓度逐渐升高,析氢反应电位逐渐正移,表明析氢变得容易,这主要是因为H2SO4浓度升高使溶液中参与析氢反应的H+浓度升高,有利于析氢过程。50 g/L H2SO4溶液中引入12.55 g/L Mn2+后,阴极析氢电位明显正移,主要是由于Mn2+的引入提高了溶液的导电性,从而促进了析氢过程。进一步对比±2.5 mA/cm2下槽压(阴阳极电位之和)的变化情况,发现随着H2SO4浓度从10 g/L升高到150 g/L,电解槽压呈现逐渐上升趋势(由2.42 V升至2.51 V),此分析结果进一步表明pH值太小不利于Mn2+的电化学氧化。考虑到Mn2+→ MnO2定向、高效转化对电解液导电性和pH值的要求(图1),后续实验选定H2SO4浓度为50 g/L。
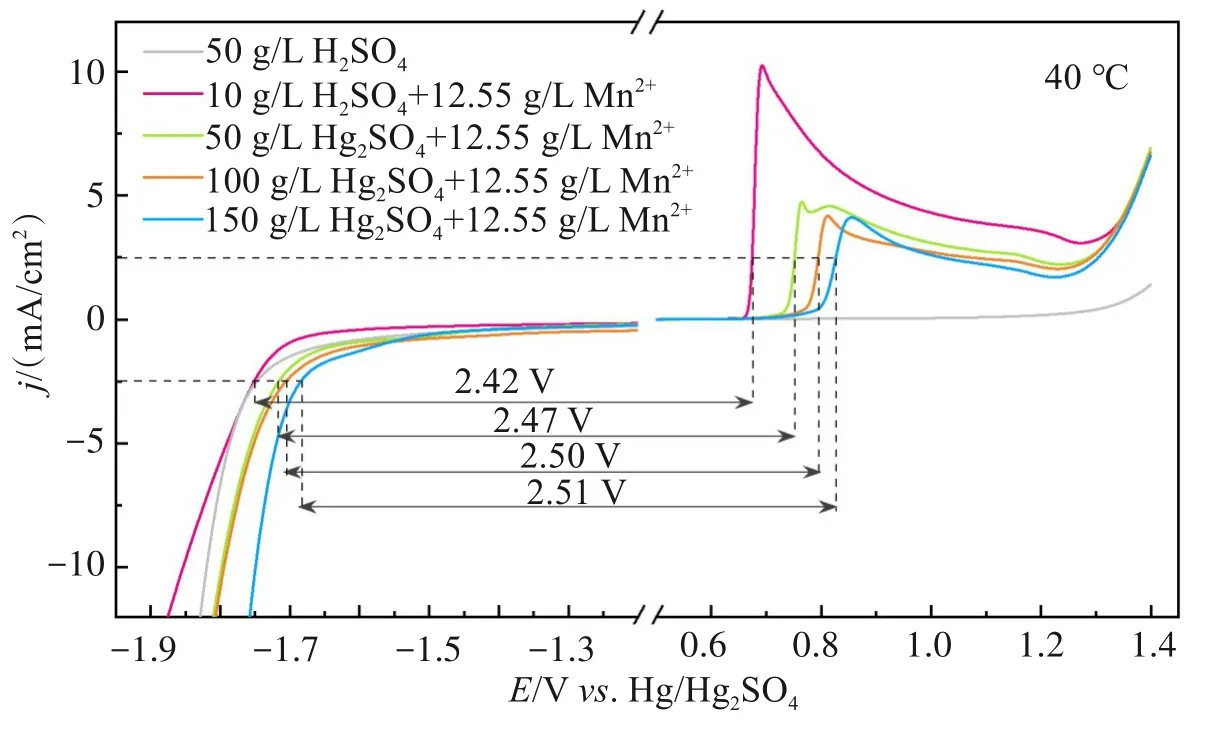
图2 不同H2SO4浓度下阴极析氢和阳极Mn2+氧化动电位极化曲线Fig.2 Potentiodynamic polarization curves of cathodic hydrogen evolution and anodic Mn2+oxidation in different concentrations of H2SO4
2.2 电化学分析
2.2.1 Mn2+的氧化还原行为
在50 g/L H2SO4+12.55 g/L Mn2+溶液中,采用CV和LSV测试进一步探索了Mn2+的氧化还原行为,如图3所示。H2SO4溶液中加入12.55 g/L Mn2+后,CV曲线显示出一对明显的Mn2+氧化还原峰(图3(a)),其中,氧化峰对应Mn2+→ MnO2的转化(式(2)),还原峰对应MnO2产物的还原。Mn2+氧化反应的起始电位约为0.74 Vvs. Hg/Hg2SO4,此数值与LSV测试结果一致(图3(b));在约0.8 Vvs. Hg/Hg2SO4时,Mn2+→ MnO2的氧化转化电流密度达到峰值,此时的电化学反应速率最快。动电位极化曲线进一步表明,当阳极电位在0.8~1.3 Vvs.Hg/Hg2SO4区间内,随着阳极电位逐渐升高,响应电流密度出现缓慢衰减,此时主要受体系中离子的传质速率限制,反应过程受到了浓差极化的影响;随着阳极电位继续增大并超过1.3 Vvs. Hg/Hg2SO4时,响应电流密度迅速增大,在此阶段,阳极上主要发生析氧和Mn2+→ MnO-4的转化。与H2SO4溶液相比,体系中引入12.55 g/L Mn2+后,1 mA/cm2下的阳极电位下降了约600 mV,表明在此酸性条件下,在热力学上,Mn2+的氧化过程优先于析氧过程。因此,通过合理控制阳极电位可避免副反应的发生,从而实现MnO2的高效电解。

图3 50 g/L H2SO4+12.55 g/L Mn2+溶液中测得(a)循环伏安曲线和(b)动电位极化曲线Fig.3 (a) Cyclic voltammetry and (b) potentiodynamic polarization curves obtained in 50 g/L H2SO4+12.55 g/L Mn2+ solution
2.2.2 Mn2+→MnO2转化的合理电位区间
上述研究结果表明,Mn2+→ MnO2的氧化转化过程存在析氧和生成MnO4-等副反应,需严格控制阳极电位以避免副反应的发生。在50 g/L H2SO4+12.55 g/L Mn2+电解液中,电解温度控制在40 ℃,采用不同电流密度进行恒流电解30 min,读取电解稳定后的阳极电位数值,得到施加电流密度与阳极电位间的对应关系,如图4(a)所示。随着施加电流密度的增加,响应阳极电位逐渐增大,当电流密度约为7 mA/cm2时,阳极电位达到Mn2+→MnO4-反应的理论值(1.51 Vvs. RHE,即0.858 Vvs.Hg/Hg2SO4,如式(3))。因此,该溶液体系下需将阳极电流密度控制在7 mA/cm2以下才能避免发生副反应。

图4 (a) 50 g/L H2SO4+12.55 g/L Mn2+电解体系中电流密度与阳极电位间的对应关系;(b)不同电流密度下电解30 min后溶液颜色变化;(c)对应溶液的紫外可见光光谱测试曲线Fig.4 (a) Corresponding relation between current density and anodic potential during Mn2+ oxidation in 50 g/L H2SO4+12.55 g/L Mn2+ solution; (b) color change of the solution after electrolysis for 30 min at different current densities;(c) corresponding UV-vis spectra
MnO4-溶液为深紫色,通过观察电解液颜色变化可初步判定MnO4-的生成情况。图4(b)显示了不同电流密度下电解30 min后溶液的颜色变化情况。与原始溶液相比,随着电流密度逐渐增大,电解后溶液颜色逐渐加深,表明生成MnO4-的副反应加剧。通过紫外-可见光光谱测试,进一步分析了溶液中MnO-4的变化情况,如图4(c)所示。401.5 nm处显示的特征峰主要对应于Mn2+;而488 nm左右的峰为MnO-4的特征峰[25],随着电解电流密度增大,此峰强度逐渐增强,表明溶液中生成了更多的MnO4-。当电流密度达到8 mA/cm2(此时响应阳极电位 > 1.51 Vvs. RHE)后,溶液变成深红色,由此可知体系中生成了大量MnO4-,因此,施加过大的电流密度不利于MnO2的高效电解。
2.2.3 Mn2+→MnO2转化的界面钝化现象
阳极电位变化和MnO2生成均会致使界面钝化,需进一步探索合适的电解电流密度以适应长周期电解过程。基于此,通过连续LSV测试研究不同温度条件下Mn2+→ MnO2转化的界面反应过程,如图5所示。50 g/L H2SO4+25 g/L Mn2+溶液中,在测试温度条件下均观察到响应电流密度逐渐下降的变化趋势,并最终稳定在一个电流平台,将其定义为极限电流密度。连续LSV扫描过程中,由于逐渐生成了MnO2膜,电化学过程受界面钝化和MnO2导电性的影响加剧,导致连续LSV扫描所响应的电流逐渐下降。所得数据表明,25 ℃下极限电流密度仅为1.86 mA/cm2,对应的槽电压为2.30 V;随着电解温度从25 ℃升至80 ℃(图5(a)—图5(d)),响应的极限电流密度显著增大到13.89 mA/cm2,表明升高电解温度可有效改善MnO2生成导致的界面钝化,提高电解反应效率。此结果与文献[26]报道的结果一致。

图5 50 g/L H2SO4+25 g/L Mn2+溶液中测得动电位极化曲线(连续扫描30次,每间隔4次取样):(a) 25 ℃;(b) 40 ℃;(c) 60 ℃;(d) 80 ℃Fig.5 Potentiodynamic polarization curves measured in 50 g/L H2SO4+25 g/L Mn2+ solution at (a) 25 ℃; (b) 40 ℃;(c) 60 ℃; and (d) 80 ℃ (Continuous scanning for 30 times with the curve taken at intervals of 4 times)
为进一步探究电极表面MnO2的生成对电解过程界面反应的影响,采用连续EIS测试监测电解反应过程中溶液/电极界面的电荷转移变化情况,如图6所示。结合CV和动电位LSV曲线结果,在室温条件下选取阳极电位为0.8 Vvs. Hg/Hg2SO4进行阻抗测试。Nyquist曲线显示,随着EIS测试次数增加(电极反应的推进),Mn2+→ MnO2转化的电荷转移电阻逐渐增大,表明电极表面MnO2的生成对反应的连续推进具有一定的阻碍作用,这主要是由于生成的MnO2导电性较差。上述结果表明,为保障Mn2+→ MnO2电解反应的连续进行,需考虑电极表面生成MnO2镀层低电导率的影响,施加过高的电流会导致电化学极化,从而引发副反应;升高温度有利于改善MnO2镀层的导电性,获得更好的电解效果。开展长周期电解时,需综合考虑电流和温度的影响。

图6 50 g/L H2SO4+25 g/L Mn2+体系100次连续EIS测试Nyquist曲线Fig.6 Nyquist plots obtained in 50 g/L H2SO4+25 g/L Mn2+ solution by 100 times of consecutive EIS tests
2.3 电解MnO2实验
结合上述电化学分析可知,Mn2+→ MnO2的氧化过程主要受界面钝化、析氧及生成MnO4-副反应等因素的影响,而这些影响因素可通过电解温度和阳极电位进行有效干预。因此,本研究进一步考察了电解温度、施加电流密度对阳极电解MnO2过程的影响。
2.3.1 电解条件探索
图7所示为50 g/L H2SO4+25 g/L Mn2+电解液中,施加不同电解温度和电流密度进行MnO2电解时响应的阳极电位变化曲线。由图7可知,电解MnO2的阳极电位经历了从初期急剧上升到中后期逐渐趋于稳定的变化过程。电解初始阶段主要为MnO2膜的生成,并逐渐覆盖在电极表面;而电解中期受电极表层MnO2导电性差的影响,电极界面电荷传递过程受阻,使得反应逐步趋缓。需要指出的是,不同电解条件下监测到的阳极电位均高于Mn2+→ MnO4-反应发生的理论值(0.858 Vvs. Hg/Hg2SO4),表明电解过程均存在副反应。

图7 不同电解温度和电流密度下电解MnO2所记录的阳极电位对随时间变化曲线Fig.7 Time-dependent curve of anodic potential pairs recorded by electrolytic MnO2 at different elctrolytic temperatures and current densities
对比不同条件下电解MnO2的阳极电流效率和阳极电位,分别如图8(a)和8(b)所示。相同电解温度下,阳极电流效率随电流密度增大而逐渐降低(图8(a))。这主要是由于MnO2的电解受极限电流密度的影响,当施加的阳极电流密度过大时,析氧和生成MnO4-等副反应加剧,导致电流效率迅速降低。此外,相同电流密度下,升高电解温度可显著提高电解效率。这与电化学分析结果一致,升高温度可有效提高阳极极限电流密度,抑制阳极极化,减少副反应的发生,从而提高电解效率。对阳极电位的变化规律进行分析发现,随电解温度升高,电解MnO2阳极电位呈现整体下降趋势(图8(b));在各电流密度下,80 ℃时响应的阳极电位总体更接近MnO4-生成的理论电位值(0.858 Vvs. Hg/Hg2SO4(1.51 Vvs.RHE))[24]。其中,6 mA/cm2条件下所得阳极电位值(0.86 Vvs. Hg/Hg2SO4)最低,表明在此条件下可有效避免副反应的发生。80 ℃时,施加6 mA/cm2电流密度获得了最佳的电解效果,MnO2电解效率达到96.6%。
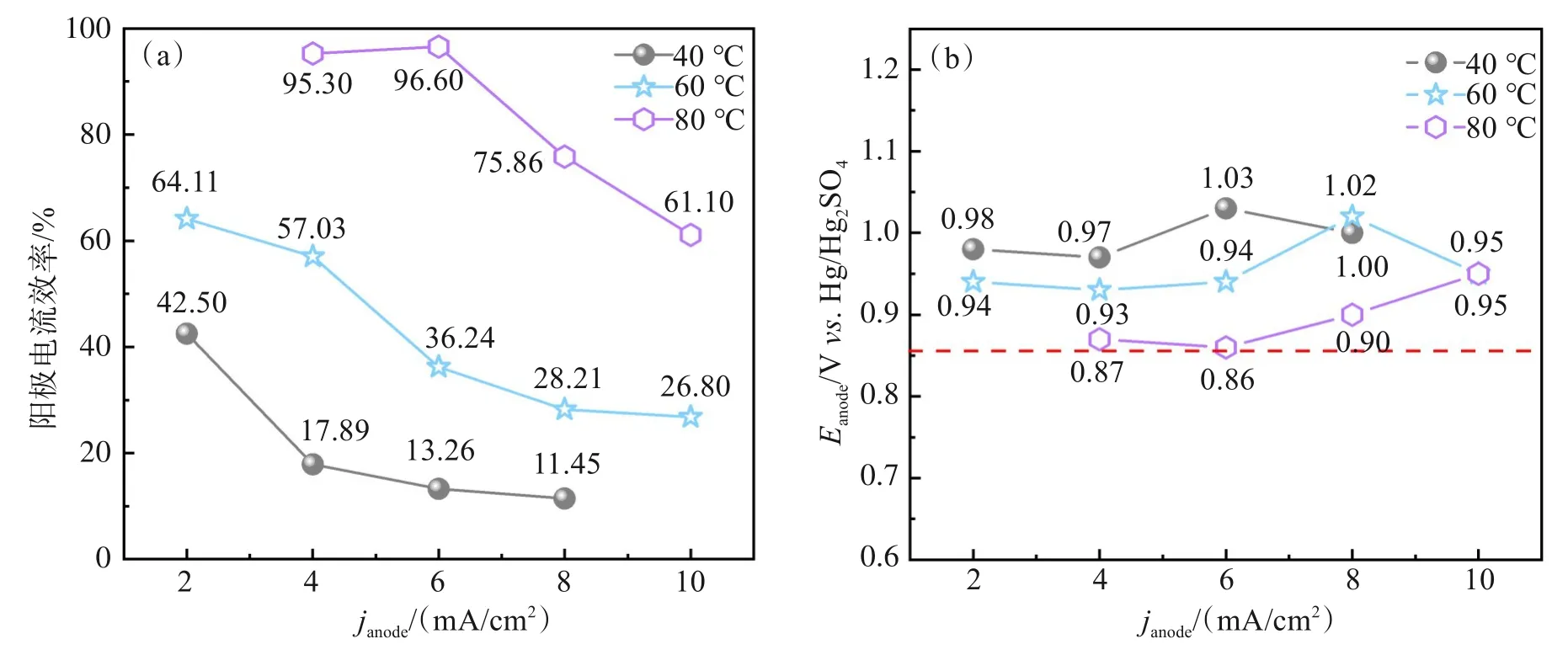
图8 不同电解温度和电流密度下电解MnO2所记录的(a)阳极电流效率和(b)平均阳极电位Fig.8 Comparison of (a) anodic current efficiency and (b) average anodic potential during MnO2 electrolysis obtained at different temperatures and current densities
2.3.2 产物物相分析
在80 ℃时不同电流密度条件下获得的产物表观和电解后溶液颜色变化如图9所示,随着电流密度增大,MnO2由致密镀层变为块状镀层,高温电解更利于剥离取样;对比电解前后溶液颜色(水溶液中MnO4-呈紫红色),各电流密度下电解前后溶液颜色无明显变化,表明电解过程中Mn2+→ MnO4-副反应未明显发生。

图9 80 ℃时不同电流密度电解MnO2的产物表观(a和b)和溶液颜色变化(c)Fig.9 Appearance of electrodeposited MnO2 (a and b) and solution color change(c) obtained at different current densities and 80 ℃
采用X-射线衍射光谱(XRD)对不同电解温度下,以6 mA/cm2进行电解时所获得的MnO2产物进行物相表征,结果如图10所示,MnO2产物均显示出宽的衍射特征峰,为α-和γ-MnO2晶型;随着温度升高,特征峰强度增强,表明升高电解温度可提高电解MnO2的结晶度。由于电解实验所用电解液均由分析纯试剂配制,本研究未对电解MnO2进行纯度分析。

图10 不同电解温度下电解产物的XRD图谱Fig.10 XRD polifiless of electrolytic products obtained at different electrolytic temperatures
3 结 论
通过系列电化学测试方法结合条件电解实验研究,系统分析了硫酸体系下Mn2+的电化学氧化行为,主要结论如下:
1) Mn2+→ MnO2电氧化反应受体系pH影响显著。体系pH > 3时不能电解得到单一的MnO2,而pH过低不利于Mn2+电氧化生成MnO2,电解体系H2SO4含量选取50 g/L (pH = 0) 为宜;酸性条件下析氧反应存在很高的过电位,在1 mA/cm2下,MnO2的生成电位较析氧电位低约600 mV,通过控制阳极电位可避免发生析氧副反应,从而实现MnO2的高效电解。
2) 动电位极化结合电化学阻抗分析表明,Mn2+→ MnO2的电氧化过程存在钝化现象,需控制合理的电流以减小极化引起的副反应;升高电解温度,可有效提高MnO2电解的极限电流密度,提升电解反应速率,避免析氧和生成MnO4-。电解温度从25 ℃升至80 ℃,Mn2+氧化的极限电流密度由1.86 mA/cm2升至13.89 mA/cm2。
3) 80 ℃下,施加6 mA/cm2电流密度电解24 h,MnO2电解效率可达到96.6%;相同条件下,40 ℃和60 ℃的电流效率仅为13.26%和36.24%;80 ℃下电解MnO2呈块状从电极上脱落,有利于产物的收集;电解MnO2主要为α-MnO2和γ-MnO2两种晶型,升高电解温度可提高产物的结晶度。
猜你喜欢
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
Water Science and Engineering(2019年2期)2019-07-24
新农民(2019年19期)2019-02-20
山东冶金(2018年6期)2019-01-28
电镀与环保(2017年5期)2017-12-19
制造技术与机床(2017年12期)2017-02-02
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
中国畜牧兽医文摘(2016年6期)2016-01-31
