
珍珠芦荟白化苗的转录组分析
2023-11-11 15:55:02陈汉鑫林艺辉马馨怡林秀芳黄婉莉
热带作物学报 2023年9期
陈汉鑫 林艺辉 马馨怡 林秀芳 黄婉莉





关键词:珍珠芦荟;白化苗;转录组;光合作用;基因表达
中图分类号:Q943.2 文献标识码:A
植物叶色白化是植物叶色突变的常见类型,在植物界普遍存在,目前已在拟南芥(Arabidopsisthaliana)、玉米( Zea mays) 、荔枝( Litchichinensis)、大麦(Hordeum vulgare)、水稻(Oryzasativa)、兰花(Cymbidium ssp.)等众多植物中发现叶片白化现象[1-7]。叶色白化产生的内在原因主要与叶绿素含量降低,光合过程受阻,叶绿体发育异常有关[8-12]。就植株本身而言,导致叶片白化的机制极其复杂,主要涉及激素失调、叶绿体形成基因突变、叶绿素合成基因突变等多种过程[1, 4]。植物叶片白化通常会导致植株光合作用受阻而影响植物的生长和发育,已被广泛应用到理论基础研究和生产实践中。如叶色突变植物是研究植物光合过程中叶绿体发育分化、叶绿素合成与降解、质核基因互作等调控机制的理想材料。在生产中,某些植物的叶片白化会带来更大的经济价值和观赏价值。安吉白茶因其叶片白化导致其氨基酸含量比普通绿茶高3~4 倍,多酚含量低于普通绿茶,提高了其营养价值和经济价值。在园林及观赏花卉植物中,特殊的叶色既丰富了园林景观的色彩,也大大提高了其观赏价值和经济价值。例如,禾本科植物竹子的自然突变体菲白竹、银丝竹,十二卷属植物黑肌玉露(Haworthia cooperivar. pilifera M. B. Bayer)的白化突变体“拉丝锦”(又名“霓虹灯”)等植物[13-14]。
芦荟是百合科芦荟属多年生常绿肉质草本植物,品种繁多,有超过500 个品种,具有药用和观赏价值[15-18]。珍珠芦荟(Aristaloe aristata)也称为木锉芦荟,属于芦荟的小型种,叶多而短,株型紧凑,主要供观赏用。珍珠芦荟叶片中含有芦荟大黄素、芦荟宁,叶肉含粘胶质丰富的多聚糖,是保健和美容护肤的佳品[19]。
珍珠芦荟白化苗,因人工繁殖的难度比较高,且叶片颜色各异,极具观赏性,在市场上比较稀有,价格比普通品种更高。鉴于珍珠芦荟白化苗存在重要的观赏价值及经济价值,为探明珍珠芦荟苗白化突变分子机制,本研究以组织培养过程中产生的珍珠芦荟白化苗突变株和正常叶色苗的叶片为研究对象,在验证其叶绿素及类胡萝卜素含量差异基础上,利用转录组高通量测序技术筛选分析珍珠芦荟白化苗的差异表达基因,挖掘相关功能基因,为进一步选育珍珠芦荟新品种提供科学的理论依据。
1 材料与方法
1.1 材料
1.1.1 供试材料 珍珠芦荟正常株(WT)及白化苗突变株(qj 和ls)由漳州市农业科学研究所组培所得。分别取生长期约为45 d 的植株叶片用于叶绿素和类胡萝卜素含量测定以及RNA 提取,每种材料3 个生物学重复。
1.1.2 主要试剂与仪器 RNA 提取试剂盒、反转录试剂盒和纯化试剂盒购自天根生化科技有限公司,Nanodrop 2000 微量分光光度计购自ThermoFisher Scientific 公司,LightCycler480 实时荧光定量PCR 仪购自瑞士Roche 公司,P8 型分光光度购自上海美谱达仪器有限公司。
1.2 方法
1.2.1 光合色素含量测定 采用分光光度计测定珍珠芦荟及其2 种突变株叶片中叶绿素和类胡萝卜素的含量[20]。称取鲜叶2.0 g 研磨,过滤,取提取液,分别在波长665、649、470 nm 下测定吸光度。根据公式分别计算出叶绿素a(Chla)、叶绿素b(Chlb)和类胡萝卜素(Caro)含量:Chla=13.95×A‒6.88×A;Chlb=24.96×A‒7.32×A;Caro=(1000×A‒2.05×Chla‒114.8×Chlb)/245。
1.2.2 RNA 提取 珍珠芦荟正常株(WT)及白化苗突变体(qj 和ls)植株叶片总RNA 提取按照RNA 提取试剂盒中的方法步骤进行,使用微量分光光度计(Nanodrop 2000)检测其纯度和浓度,将符合要求的RNA 置于‒80 ℃冰箱中保存备用。
1.2.3 文库的构建及转录组测序 为了使测序结果更稳定,本研究将3 个合格样品混合为1 个样品,并在此基础上做3 个生物学重复。使用oligo(dT)磁珠从总RNA 中富集带poly(A)的mRNA,随后采用超声处理成随机片段。以mRNA為模板,经逆转录反应合成第一链cDNA 和第二链cDNA,接着用纯化试剂盒纯化cND,最后进行末端修复、加尾和加接头,构建成cDNA 文库,基于IlluminaHiSeq TM 4000 高通量平台,以Pair-end 150 bp双端测序模式对cDNA 文库进行转录组测序。
1.2.4 Unigene 序列的获得和生物信息学分析首先获得原始读长(raw reads),进一步过滤和质控获得clean reads,随后借助Trinity 软件根据序列之间的重叠信息(kmer 长度为31 bp,kmer深度为6)组装拼接得到Unigene。将获得的Unigene 序列与Nr、SwissProt、GO、KEGG 和COG 等数据库进行BLAST 比对分析,得到芦荟基因的蛋白功能注释和分类。
1.2.5 差异表达基因鉴定与功能富集分析 采用DESeq2[21]进行基因差异表达分析。以FDR<0.05且|log2FC|>1 为标准进行差异表达基因的鉴定。其中FDR<0.05 且logFC>1 的基因为上调表达基因,FDR<0.05 且logFC<-1 的基因为下调表达基因。通过FPKM值计算定量Unigenes 的表达丰度,公式:FPKM=10×C/(N×L/1000)。通过BLASTx程序以e-value<1e-5 为阈值在Nr 数据库、Swiss-prot 蛋白数据库、KEGG 数据库和KOG 数据库对Unigenes 注释。基于Nr 注释,借助Blast2GO 软件进行GO 注释分析[22]。通过WEGO软件对Unigenes 分类[23]。GO 与KEGG 富集分析是利用超几何检验进行P 值计算并进行Bonferroni 校正,以q-value<0.05 为标准筛选出差异表达基因显著富集的GO 条目或pathway。
1.2.6 DEGs 的qRT-PCR 验证 采用TaKaRa 公司的PrimeScriptTM 逆转录试剂盒进行逆转录合成cDNA。采用TaKaRa 公司的SYBR Premix ExTaqTM 进行Real-time PCR,反应条件为:98 ℃预变性2 min;95 ℃变性10 s,60 ℃退火15 s,68 ℃延伸30 s,35 个循环。内参选用珍珠芦荟actin,利用Primer 5.0 对验证基因的CDS 区域设计特异性引物,引物详细信息见表1。采用2–ΔΔCT 法计算珍珠芦荟不同材料间差异基因的相对表达量。比较转录组水平的FPKM 值和荧光定量PCR 的相对表达量,验证转录组数据筛选DEGs 的可靠性。
1.3 数据处理
使用Excel 2016 和Origin 8.0 软件进行数据整理和制图,采用Duncan’s 新复极差法检验。
2 结果与分析
2.1 珍珠芦荟白化苗突变株及其光合色素含量分析
与正常苗对比,白化苗突变株ls 外部色泽呈现白绿相间,白化苗突变株qj 外部色泽呈现淡淡的绿白色,而正常叶色苗呈现均匀绿色(图1)。对珍珠芦荟3 种材料叶片的光合色素叶绿素a、叶绿素b 和类胡萝卜素含量测定结果表明,正常植株叶片中叶绿素a、叶绿素b 和类胡萝卜素含量极显著高于白化突变体(ls 和qj)。与正常苗叶片相比,突变体ls 叶片的叶绿素a、叶绿素b和类胡萝卜素含量分别降低63.8%、61.8%和61.1%,突变体材料qj 叶片的叶绿素a、叶绿素b 和类胡萝卜素含量分别降低73.4%、76.0%和53.1%(图2)。
2.2 转录组数据分析
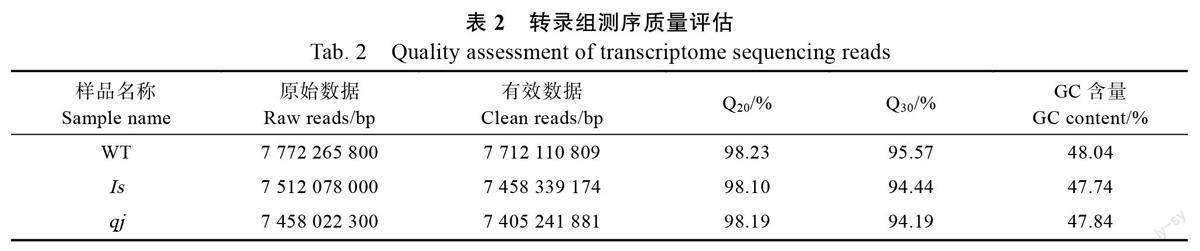
本研究共获得原始数据(raw reads)68.23 Gb,经过数据过滤和质控得到有效数据(clean reads)67.72 Gb,GC 含量介于47.63%~48.25%之间,Q20平均值超过98%,Q30 平均值超过94%(表2)。经过组装,一共得到122 665 条Unigenes,其中,最长长度为24 511 bp,最短长度为201 bp,平均长度为822 bp,Unigene N50 数量为19 100,N50长度为1443 bp。不同长度Unigenes 分布比例显示,长度在200~500 bp 的Unigene 占55.9%,长度超过2000 bp 的占9.36%。上述结果表明,测序结果良好,各项质控指标达到要求,可用于后续分析。
2.3 Unigene 功能注释 将组装得到的122 665条Unigenes 序列与相关数据库进行比对,其中,51 798 条Unigenes 在Nr 数据库(non-redundantprotein database)中获得注释,占42.2%;35 338条Unigenes 注释到SwissProt 数据库,占28.8%;30 666 条Unigenes 在COG 数据库中获得注释,占25.0%;29 046 条Unigenes 在GO 数据库中获得注释,占23.7%;45 496 条Unigene 在KEGG数据库中获得注释,占37.1%(图3)。
将比对到Nr 数据库的序列进一步分析,结果表明,珍珠芦荟与芦笋(Asparagus officinalis)匹配比例最高,为8822 条,占到可匹配序列的7.19% , 接下来依次为蒺藜苜蓿( Medicagotruncatula,5.89%)、棕榈(Elaeis guineensis,1.93%)、葡萄(Vitis vinifera,1.68%)、海枣(Phoenixdactylifera,1.57%)、金杜鹃(Rhodamnia argentea,1.29%)、石斛(Dendrobium catenatum,1.22%)、黄花蒿(Artemisia annua,1.14 %)(图4)。
2.3 差异表达基因的GO富集分析
以FDR<0.05 且|log2FC|>1 为条件进行差异表达基因的筛选。结果表明(图5),与正常植株WT 的叶片相比,白化苗突变株ls 叶片中有914个差异表达基因,其中453 个上调,461 个下调(WT-vs-ls);白化苗突变株qj 叶片中有1851 个差异表达基因,其中868 个上调,983 个下调(WT-vs-qj)。
为了研究珍珠芦荟白化苗突变株的DEGs 涉及的生物学功能,将白化植株ls 和qj 中的DEGs分别进行了GO 功能富集分析。由图6A 和图6B可知,ls 和qj 的DEGs 均被富集到生物过程(biological process, BP)、细胞组分(cellularcomponent, CC)和分子功能(molecular function,MF)3 個功能类别。在生物过程中,ls 的DEGs显著富集在色素积累(pigment accumulation)、组织中的色素积累(pigment accumulation in tissues)、细胞壁组成或生物发生(cell wall organizationor biogenesis ) 、发育性生长( developmentalgrowth)等;qj 的DEGs 则显著富集在细胞壁组成或生物合成(cell wall organization or biogenesis)、内源性刺激反应(response to endogenousstimulus)和色素聚集(pigment accumulation)等生物过程。在细胞组分中,ls 的DEGs 显著富集在细胞壁(cell wall)、细胞外周(cell periphery)、细胞外区域(extracellular region)等;qj 的DEGs则显著富集在细胞外周(cell periphery)、细胞壁(cell wall)和外部封装结构(external encapsulatingstructure)。在分子功能中,ls 显著富集在水解O-糖基化合物水解酶活性(hydrolase activity,hydrolyzing O-glycosyl compounds)、作用于糖基键的水解酶活性(hydrolase activity, acting onglycosyl bonds)和酶抑制剂活性(enzyme inhibitoractivity);qj 的DEGs 则显著富集在在水解O-糖基化合物水解酶活性(hydrolase activity, hydrolyzingO-glycosyl compounds)、作用于糖基键的水解酶活性(hydrolase activity, acting on glycosylbonds)和氧化还原酶活性(oxidoreductaseactivity)。
2.4 差异表达基因的KEGG富集分析
为研究白化苗突变株ls 和qj 的代谢通路情况,本研究对ls 和qj 的DEGs 分别进行KEGG 富集分析。结果表明,ls 的差异表达基因显著富集在植物-病原互作(plant-pathogen interaction)、代谢通路(metabolic pathways)、MAPK 信号通路-植物(MAPK signaling pathway-plant)、二萜生物合成(diterpenoid biosynthesis)及次生代谢物生物合成(biosynthesis of secondary metabolite)等通路(图7A);而qj 的DEGs 则显著富集在核糖体(ribosome)、脂肪酶延伸(fatty acid elongation)、次生代谢物生物合成(biosynthesis ofsecondary metabolite ) 、苯丙烷生物合成(phenylpropanoid biosynthesis)和类黄酮生物化成(flavonoid biosynthesis)等通路(图7B),2个突变体ls 和qj 均在次生谢物生物合成中差异表达基因存在显著富集现象。
2.5 参与光合作用相关基因的筛选及表达分析
基因porA 和Cab13 与叶绿素的合成紧密相关,其在2 个突变体中均呈现出显著的下调表达。在与光合作用相关基因(如光合途径、天线蛋白、叶绿素合成)中,以FDR<0.05 且|log2FC|>1 为阈值,从突变体ls 中选取了7 个基因,在突变体qj中选取了22 个基因进行分析。结果表明,与正常植株WT 相比,在突变体ls 中,呈现显著下调的基因有原叶绿素酸酯氧化还原酶A(porA)和叶绿素a/b 结合蛋白(cab13)(图8);在突变体qj 中,呈现显著下调的基因有moda 和porA(图8)。与叶绿素合成紧密相关的基因porA 和Cab13在2 个突变体中均呈现出显著的下调表达。
为验证转录组数据可靠性,采用qRT-PCR对与叶绿素合成的相关基因porA、cab13 和moda 表达情况进行验证,结果表明(图9),与正常植株WT 相比,白化苗突变株ls 和qj 的porA、cab13 和moda 的表達均显著下调,该数据与转录组中FPKM 值的变化数据一致,说明该转录组数据可靠。
3 讨论
白化突变虽一般为致死性突变,许多植株在幼苗期就会死亡,因此在实际应用中受到了限制,但园艺植物的白化突变体却有极高的观赏价值,且白化现象也具有一定的理论价值。白化突变体可以用来标记性状,也可以作为特殊的种质资源来研究与叶绿体发育和叶绿素合成的相关基因的调控机制。高等植物叶片中叶绿素a、叶绿素b和类胡萝卜素等三大类光合色素含量和分布决定了其颜色[24-27]。本研究通过测定珍珠芦荟正常植株(WT)和白化苗突变株(ls 和qj)的光合色素表明,白化苗突变株的叶绿素a、叶绿素b 和类胡萝卜素的含量均远远低于正常植株。
许多研究表明,植物叶绿素的生物合成是一个高度有序的生理生化过程,20 多个基因编码的16 种酶参与该过程,当此过程发生障碍,则前体物质会积累,进而导致其含量增加,其后的物质含量会迅速下降,从而产生颜色变异的叶片;类胡萝卜素广泛存在于植物的绿色组织中,它可以保护叶绿素免受光氧化。在合成胡萝卜素和叶黄素的过程中,相关基因发生突变,也能引起植物白化现象的产生,因此,叶色白化突变体形成的内在原因主要是叶绿素合成代谢受阻导致的[2, 4, 5]。叶绿素的合成与代谢是一个复杂调控过程,涉及许多酶促反应,其中每一步酶促反应的变化都将影响叶绿素的最终合成而使叶色发生变化甚至导致植株死亡。已有研究结果表明,与叶绿素合成和降解紧密相关基因的异常表达是促使叶片白化现象的主要原因[28]。
在本研究中,对叶色正常和白化的珍珠芦荟进行了转录组测序,结果表明在白化苗突变株ls叶片中共筛选到914 个差异表达基因(DEGs),其中453 个上调,461 个下调;在白化苗突变株qj 叶片中筛选到1851 个差异表达基因,其中868个上调,983 个下调。GO 功能富集分析显示,ls和qj 的差异表达基因均显著富集在色素积累(pigment accumulation)的生物过程中;在分子功能则均显著富集在水解O-糖基化合物水解酶活性(hydrolase activity, hydrolyzing O-glycosylcompounds ) 和作用于糖基键的水解酶活性(hydrolase activity, acting on glycosyl bonds)中。在对差异基因的KEGG 富集分析发现,ls 和qj均显著富集在次生代谢物合成等通路中,而与光合作用相关途径的差异基因未获得显著富集。在对光合色素合成相关基因的表达情况分析发现,这些基因在2 个白化突变株中呈现出显著差异表达,其中与叶绿素合成的关键基因除porA、cab13和moda 在白化突变株呈现显著下调外,sgr 等其他搜寻获得的基因呈现上调表达;与类胡萝卜素降解相关的基因9-顺式-环氧类胡萝卜素双加氧酶(nced)表达呈现显著上调表达,这与光合色素含量的测定结果相一致。研究表明,呈现下调表达的基因porA、cab13 和moda 为叶绿素合成相关酶编码基因,这些基因的突变或下调表达会影响植物叶素绿的合成。
porA 为原叶绿素酸酯氧化还原酶,它催化底物原叶绿素的还原,最终形成叶绿素。porA 为2个突变株共同下调表达的基因,porA 基因下调会导致原叶绿素酸酯合成叶绿素酸酯的酶促反应受到影响,进而影响2 个突变株的叶绿素合成,因此推测该基因下调对珍珠芦荟白化苗形成起着重要的作用,这与左朋[29]研究中利用过表达porA、porB 基因促进叶绿素的合成的研究结果相符合。研究显示porA-1 突变体以及porA RNAi 植株会呈现出严重的光合自养生长缺陷[30]。在暗形态发生中,缺失porA 会导致初级质体基粒体积和数量减少,以及光活性的植物原叶绿素酸酯转化的减少[30]。此外,porA 基因对质体发育中膜的超微结构以及正常生化反应是至关重要的[30];而在呈现上调表达的基因中,sgr 编码一个Mg2+去螯合酶,参与到叶绿素的降解过程,sgr 在水稻中过量表达能促进叶绿素分解,减少正常生长叶片中的基粒类囊体片层数量和叶绿素含量[31]。番茄、水稻、鳄梨、葡萄及草莓等植物中nced 基因主要通过参与到脱落酸生物合成过程,导致植物体内脱落酸含量变化,使植物器官呈现不同颜色[32]。
本研究以珍珠芦荟及其白化突变株为研究对象,通过转录组学分析挖掘出与叶片白化形成相关的关键功能基因,研究结果为揭示珍珠芦荟组培苗白化突变体形成可能包含的分子机制奠定了理论基础。
猜你喜欢
科学(2022年4期)2022-10-25 02:43:00
中国中药杂志(2017年2期)2017-03-25 17:23:23
中国中药杂志(2017年1期)2017-03-06 21:20:38
中国中药杂志(2016年22期)2017-02-13 17:06:29
中国科技博览(2016年24期)2016-12-28 23:53:07
中国实用医药(2016年30期)2016-12-28 16:54:00
中国民族民间医药·上半月(2016年10期)2016-11-19 11:08:04
中国科技博览(2016年22期)2016-11-01 13:58:50
Coco薇(2016年5期)2016-06-03 09:17:41
考试周刊(2016年6期)2016-03-11 08:14:32
