
安徽地区克氏原螯虾群体的遗传多样性和遗传结构
2023-11-05 08:46崔文涛邹宇凡白志毅王志炎李典中
水产学报 2023年9期
崔文涛,邹宇凡,白志毅,2*,王志炎,李典中
(1.上海海洋大学,农业农村部淡水水产种质资源重点实验室,上海 201306;2.上海海洋大学,上海市水产养殖工程技术研究中心,上海 201306;3.芜湖盛典休闲生态园有限公司,安徽 芜湖 241200)
生物的遗传多样性主要体现在不同个体或群体间DNA 和蛋白质序列的遗传距离和差异上,其不受时间的推移而增加,主要受限于功能和表观遗传的复杂性[1]。遗传多样性通常通过限制性片段长度多态性(RFLP)、扩增片段长度多态性(AFLP)、随机扩增多态DNA (RAPD)、微卫星(SSR)和单核苷酸多态性(SNP)等分子标记来评估,微卫星也称为SSRs,是一种在真核生物基因组中具有高度变异的简单重复DNA 序列[2]。由于其符合孟德尔的遗传规律,具有高度多态,共显性,且高度可复制,易于操作[3]等优点,因此,通过SSR 获得的遗传多样性数据可用于调查的物种遗传变异性,且为防止物种的遗传多样性随着时间推移而丧失提供了理论参考[4]。SSRs 也被广泛应用于水产动物的遗传连锁图谱构建、数量性状基因座(QTL)作图、群体遗传分析、亲子鉴定和分子标记辅助选择(MAS)等[5-6]。
克氏原螯虾(Procambarus clarkii)俗称淡水龙虾,其具有适应性广、繁殖力强、生长快、肉质鲜美及营养丰富等特点,广受国内外养殖者和消费者青睐。克氏原螯虾原产于美国东南部和墨西哥东北部[7],后经日本引入我国[8],已在我国各地广泛分布并形成了不同的地理种群。国外学者均已先后对其国内克氏原螯虾的种群扩散过程、机制和种群遗传结构的做了大量研究工作[9-11]。在国内,Yue 等[12]等利用微卫星标记分析了6 个地区的克氏原螯虾遗传多样性和种群结构。李艳和[13]对我国的35 个群体以及1 个美国种群、1 个日本种群进行群体遗传多样性和种群遗传结构分析。邢智珺等[14]分析了江苏8 个克氏原螯虾群体遗传多样性和遗传结构。上述研究结果表明,尽管我国克氏原螯虾遗传多样性低于美国、日本群体,但整体遗传多样性水平较高。
克氏原螯虾的稻虾种养面积、水产品产量分别占全国稻渔综合种养总量的47.70%和60.84%,为最大规模的稻渔综合种养模式[15]。根据《中国小龙虾产业发展报告(2020)》[16],2019 年安徽地区的克氏原螯虾产量达34.98 万t,位居全国第二。随着我国克氏原螯虾养殖规模扩大,成虾规格、品质带来的市场价格差距加剧了养殖业的竞争。而克氏原螯虾养繁一体的养殖模式及捕大留小的苗种供给方式,不利于亲本留种和良种选育,导致克氏原螯虾种质的衰退及抗病力低等问题,严重制约着我国克氏原螯虾养殖业的进一步发展[17]。目前对安徽地区克氏原螯虾养殖群体的遗传多样性评估鲜有报道,因此有必要对安徽克氏原螯虾养殖群体进行遗传多样性的评估。本研究利用10对微卫星标记分子对安徽(3 个人工养殖群体和2个野生群体)、湖北(1 个人工养殖群体)和江苏(3个人工养殖群体)共9 个克氏原螯虾群体的遗传结构进行分析,旨在了解安徽不同地区人工养殖的克氏原螯虾群体间的遗传差异,为生产养殖提供理论指导。
1 材料与方法
1.1 样品采样和DNA 的提取
2020 年分别采集了芜湖(WH)、宣城(XC)、合肥(HF)、监利(JL)、建湖(JH)、滆湖(GH)、兴化(XH) 7 个地区的稻渔综合种养示范区人工养殖群体和铜陵(TL)、马鞍山(MAS) 2 个地区的野生群体(图1),共270 尾克氏原螯虾样品用于实验研究。将所有克氏原螯虾的尾壳去除,露出白色肌肉,然后收集肌肉组织,并用无水乙醇固定,然后按照制造商的说明,使用海洋动物组织基因组试剂盒[天根生化科技(北京)有限公司]进行总DNA 提取,利用分光光度计测定其浓度和纯度,并于-20 °C 低温冰箱中保存备用。本研究获得了上海海洋大学实验动物管理和使用伦理委员会批准(SHOU-DW-2019-003),实验过程中操作人员严格遵守上海海洋大学伦理规范,并按照上海海洋大学伦理委员会制定的规章制度执行;所有实验均按照当地的研究动物使用准则进行,并经伦理委员会批准。
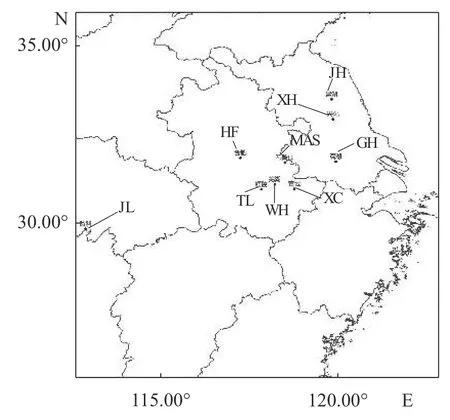
图1 9 个克氏原螯虾群体采样地点JL.监利群体,JH.建湖群体,GH.滆湖群体,XH.兴化群体,TL.铜陵群体,MAS.马鞍山群体,WH.芜湖群体,XC.宣城群体,HF.合肥群体;下同。Fig.1 Sampling sites of 9 P.clarkii populationsJL.Jianli populations,JH.Jianhu populations,GH.Gehu populations,XH.Xinghua populations,TL.Tongling populations,MAS.Maanshan populations,WH.Wuhu populations,XC.Xuancheng populations,HF.Hefei populations;the same below.
1.2 引物的合成
为了节约实验成本,本研究参照了Schuelke[18]TP-M13-SSR 技术,把M13 正向引物的5′端带上荧光标记(表1),选用10 对已发表、扩增条件较好、多态性高的克氏原螯虾SSR 引物[10,14,19]的反向引物和M13 引物相连作为M13-SSR 的正向引物,其正常的正向SSR 引物作为M13-SSR 的反向引物(表2);M13-SSR 引物及5′端带有荧光标记的M13 正向引物引物均由生工生物工程(上海)股份有限公司合成。
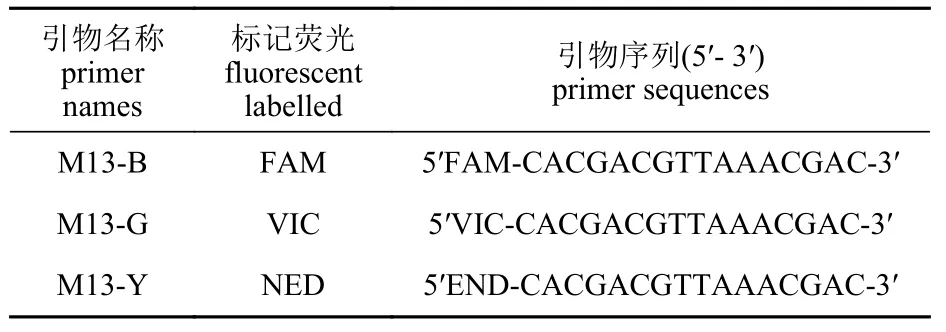
表1 5′端带有荧光标记的M13 正向引物及序列Tab.1 The M13 forward primer fluorescent-labelled at 5′end
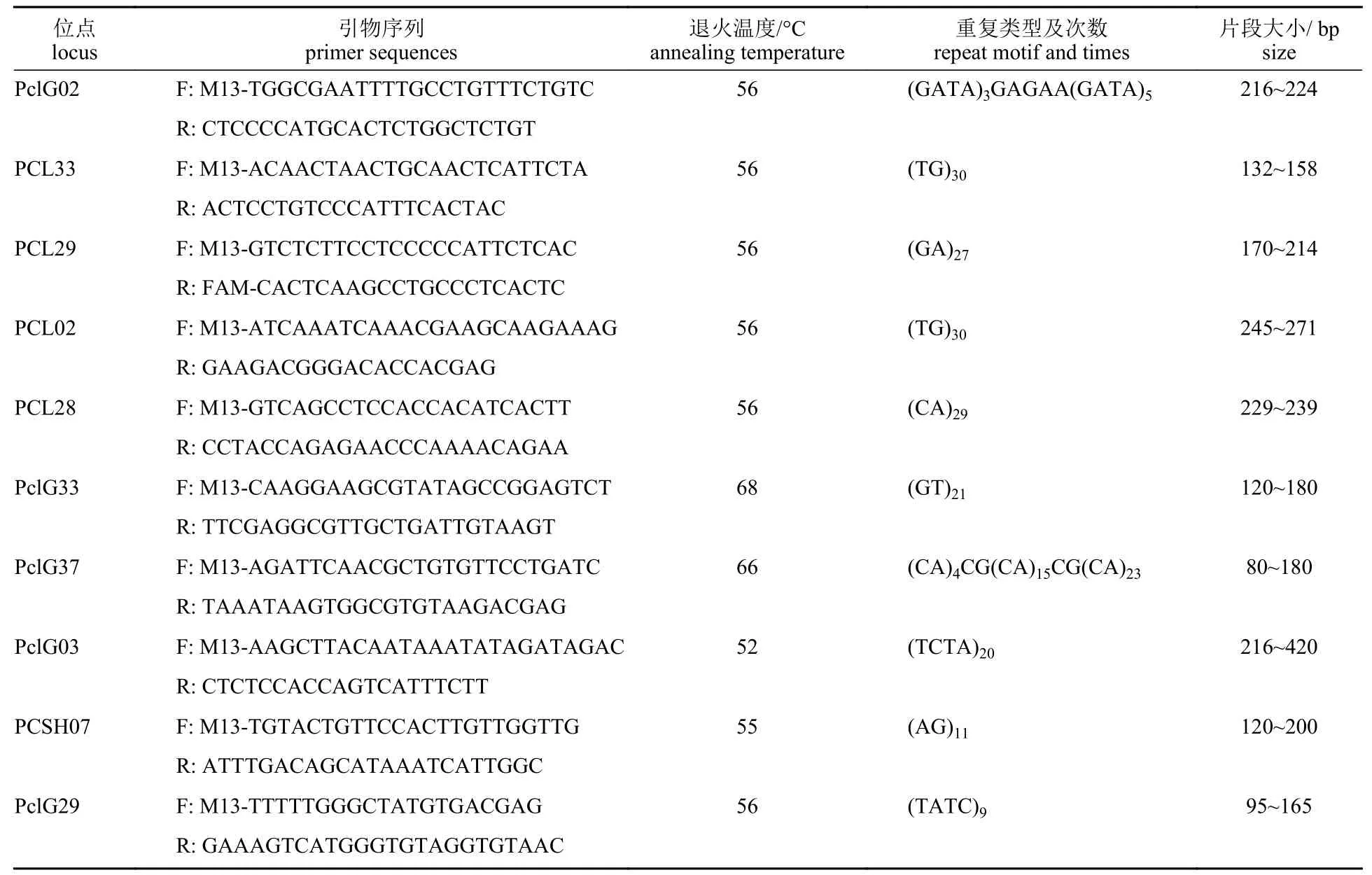
表2 克氏原螯虾10 对微卫星引物Tab.2 Primers of 10 microsatellites in P.clarkii
1.3 PCR 扩增与数据分析
PCR 反应分2 步进行。第1 步把SSR 反向引物与M13 的正向引物相连扩增为M13-SSR 引物,第2 步SSR 扩增产物的荧光标记,使用M1-SSR3引物与SSR 正向引物扩增获得的PCR 产物与表1中M13 荧光引物再进行扩增,所采用的PCR 反应体系均经过了优化。在筛选引物时,先用PAGE 检测引物的扩增效果,然后选取扩增效果理想的扩增产物交由生工生物工程(上海)股份有限公司进行分型(图2)。
采用软件POPGEN 3.2 统计群体的遗传多样性参数;根据Botstein 公式[20]计算Hardy-Weinberg 遗传偏离指数(D)[21],采用Bonferroni 方法校正显著性标准;群体遗传分化指数(Fst)及基因流(Nm)利用软件CERVUS 3.0 统计并进行分子方差分析(AMOVA);Structure 2.3[22-23]等软件对群体的遗传结构进行统计和分析;基于遗传距离用Mega 4.0 软件构建UPGMA 系统进化树。
2 结果
2.1 种群遗传多样性分析
10 对微卫星引物扩增结果显示,等位基因数(Na)为3~33 个(平均15.70 个),有效等位基因数(Ne)为1.50~13.89 (平均4.76 个),观测杂合度(Ho)为0.12~0.91,期望杂合度(He)为0.33~0.93;各群体的多态性信息含量(PIC)为0.31~0.92,除了PCL28 位点为中度多态性外,其他9 个位点均属于高度多态性位点(PIC>0.5) (表3),可用于微卫星遗传结构分析。
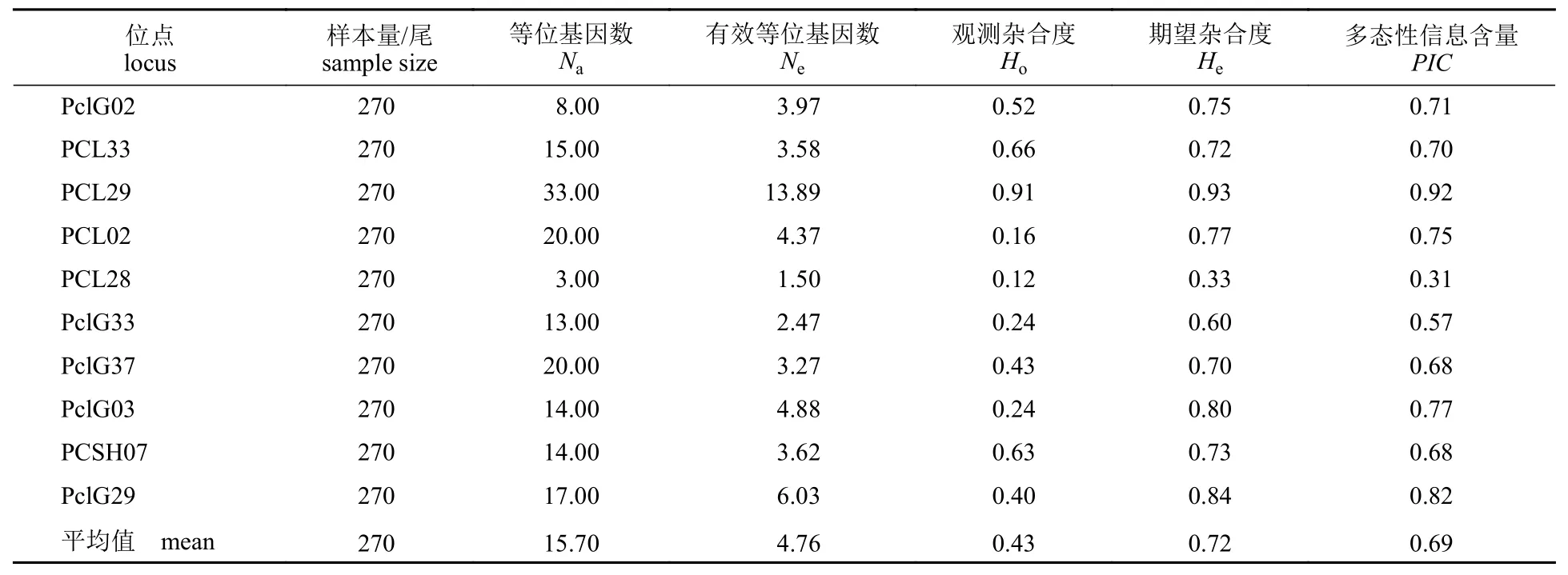
表3 克氏原螯虾15 个微卫星位点的遗传多样性参数Tab.3 Statistics of number of effective alleles,expected and observed heterozygosity and polymorphism information content for microsatellite loci of P.clarkii
9 个克氏原螯虾群体(270 尾)的遗传多样性如表4 所示,总体上,9 个群体均具有较高的遗传多样性。群体水平遗传多样性参数比较发现,XC 群体在等位基因数(Na=7.70)、有效等位基因数(Ne=4.40)、观测杂合度(Ho=0.50)、期望杂合度(He=0.74)和多态性信息含量(PIC=0.67)均最高,与其他群体间均存在显著差异(P>0.05),而JL 群体的遗传多样性最低(PIC=0.48),其群体的遗传多样性有待提高。总体上9 个群体均具有较高的遗传多样性,且安徽地区人工养殖群体的遗传多样性普遍高于其他几个群体。
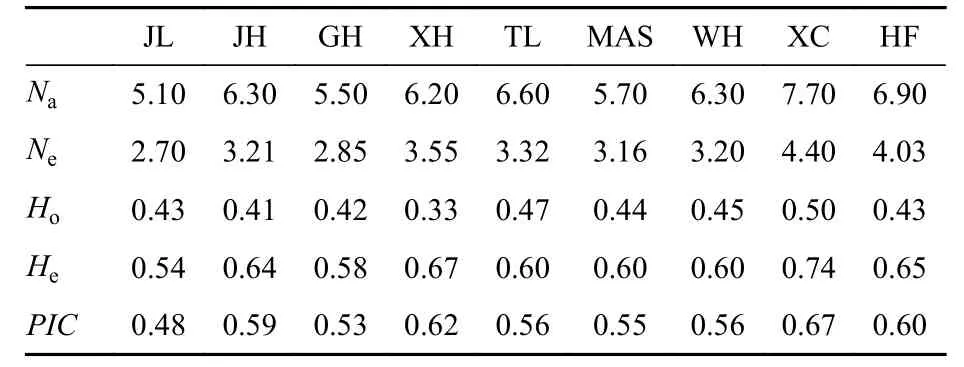
表4 克氏原螯虾9 个群体的遗传多样性参数Tab.4 Summary statistics of genetic diversity of nine P.clarkii populations
利用Hardy-Weinberg 平衡对9 个群体中所有位点基因平衡状态进行检验,9 个群体各位点平均遗传偏离指数为-0.568~-0.178,经邦弗朗尼校正,9 个群体的大多数位点均显著偏离Hardy-Weinberg 平衡(表5),与其他群体相比,HF 群体的偏离程度最高,JL 和XH 群体的偏离程度最低。此外,人工养殖群体和野生群体均表现出杂合子缺失现象,由于其具有较强的繁殖力,亲缘关系较近的后代极易发生近亲繁殖,说明近亲繁殖是群体间偏离Hardy-Weinberg 平衡的主要因素,为安徽地区克氏原螯虾人工养殖群体的遗传改良提供参考。
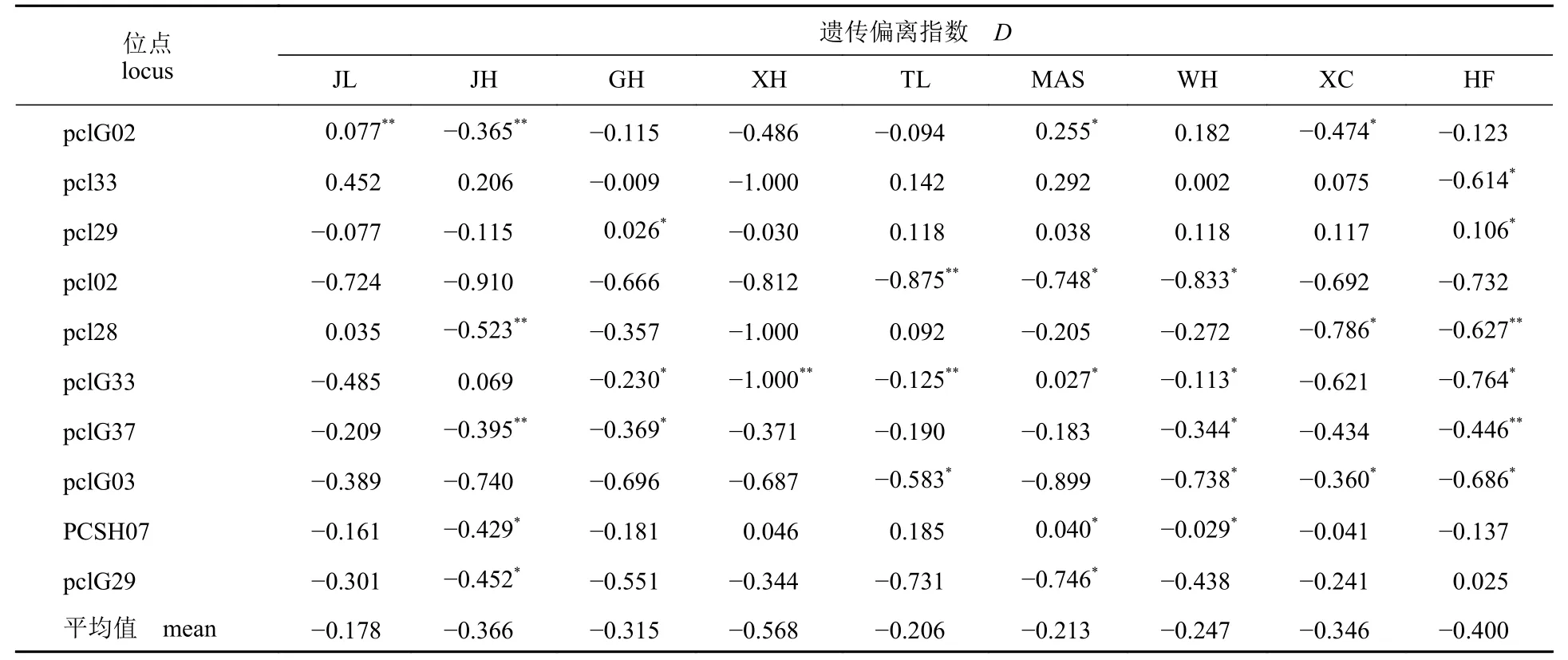
表5 克氏原螯虾9 个群体遗传偏离指数(D)Tab.5 Deviation (D) assessed for the nine populations of P.clarkii
2.2 种群遗传分化分析
克氏原螯虾9 个群体基因流(Nm)及群体遗传分化指数(Fst)显示,各群体间基因流为1.028~10.299,不同群体之间存在着广泛的基因交换,GH 和JH 群体间的基因交流最广泛(Nm=10.299)。群体遗传分化指数为0.024~0.196,其中除了XC群体的分化程度较低(Fst<0.05),HF 群体与其他群体的Fst均表现出高度分化(Fst>0.15) (表6)。
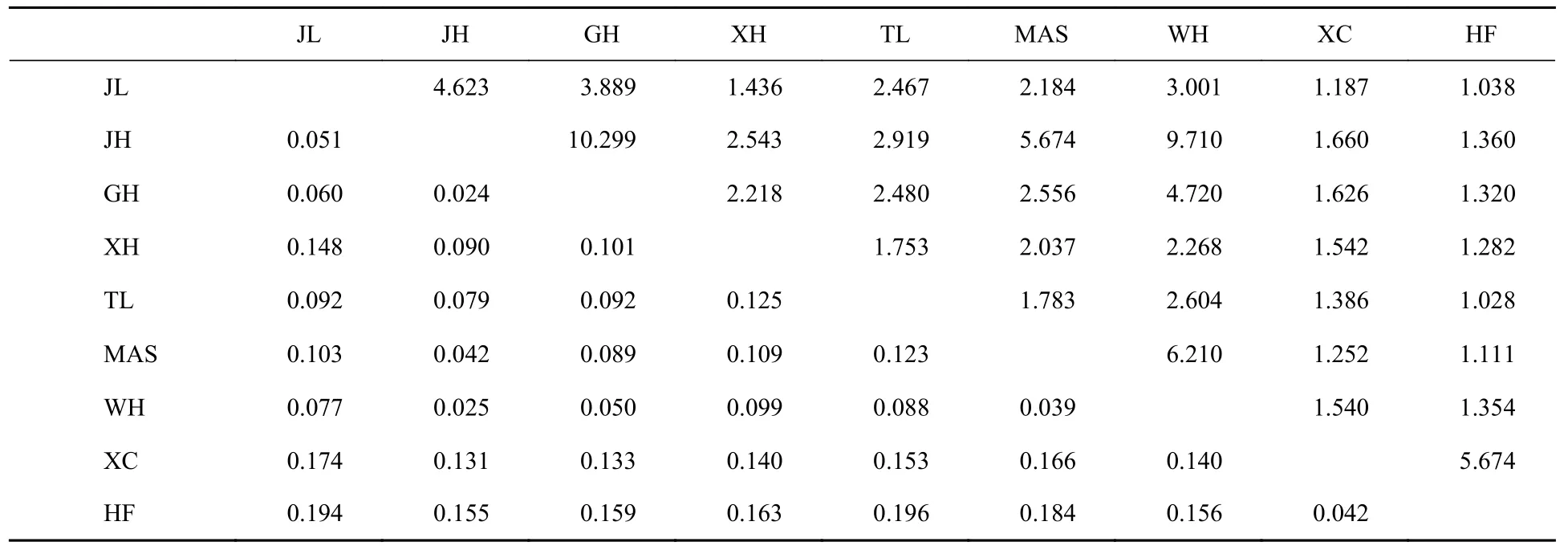
表6 克氏原螯虾9 个群体基因流(Nm,对角线上)和遗传分化系数(Fst,对角线下)Tab.6 Gene flow (Nm,above diagonal) and genetic diversity (Fst,below diagonal) in nine P.clarkii populations
9 个克氏原螯虾群体遗传相似系数和遗传距离结果显示,各群体间遗传相似系数(I)为0.078~0.939,遗传距离(DA)为0.064~0.746;WH 群体与JH 群体的遗传距离最小,遗传相似度最大。而JH 群体与XH 群体的遗传距离最大,遗传相似度最小(表7)。AMOVA 分析结果表明,88.58%的遗传变异来自群体内变异,而其余的(11.42%)来自群体间的变异,群体间遗传分化达到极显著水平(P<0.01) (表8)。

表7 克氏原螯虾9 个群体间遗传相似系数(I,对角线上)及遗传距离(DA,对角线下)Tab.7 Genetic similarity indices (I,above the diagonal) and genetic distances (DA,below the diagonal) among nine populations of P.clarkii
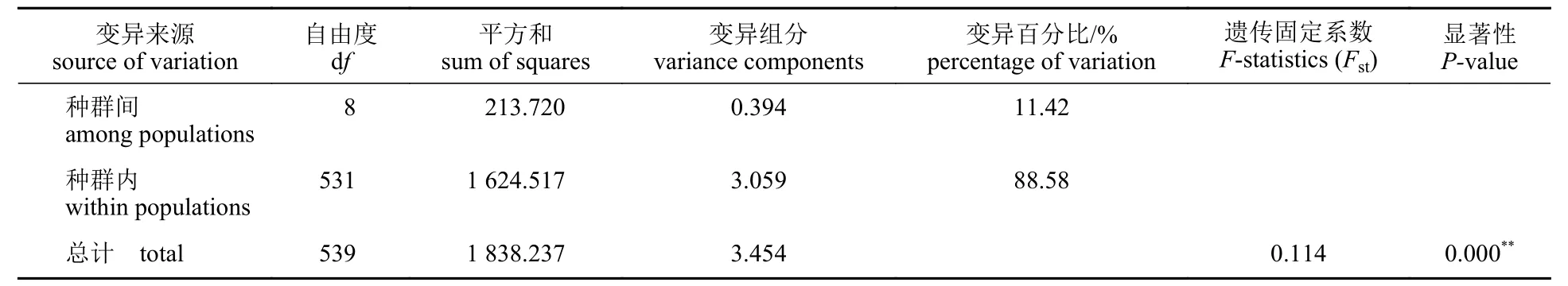
表8 安徽省克氏原螯虾群体分子方差分析Tab.8 AMOVA analysis among P.clarkii populations in Anhui Province
基于Nei 氏遗传距离对克氏原螯虾9 个群体构建UPGMA 系统树,结果显示,9 个群体可分为5 组,即WH、GH、MAS 和JH 群体聚为一组,XC 和HF 群体同属于一组,而TL、XH 及JL 群体分别自成一组(图3)。
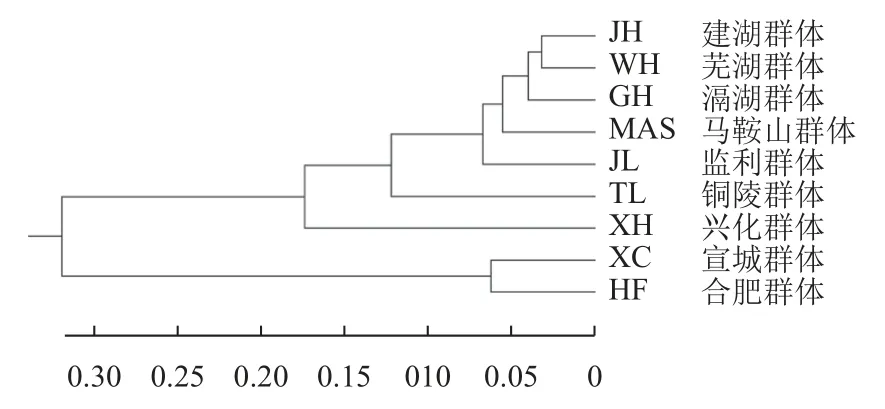
图3 基于Nei 氏遗传距离的UPGMA 聚类树Fig.3 UPGMA clustering tree based on Nei’s genetic distance
2.3 种群遗传结构分析
本研究根据K值对应参数的趋势分析,发现K=5 时出现折点(图4,图5),说明最有可能的亚种群数为5。图5 给出了K=3、K=4 和K=5 的遗传结构图。在养殖群体中XC 和HF 2 个养殖群体的大多数个体被分配到相同的遗传群的中,这表明它们具有较高的遗传相似性;而其他养殖群体的一些个体较混杂。在野生群体中,TL 群体的个体遗传结构相对单一,与其他群体的个体有明显区别。
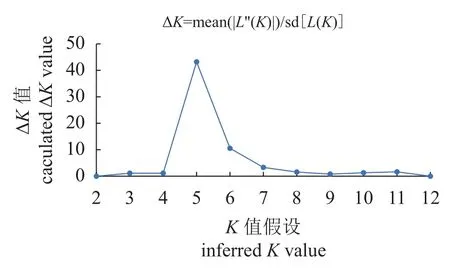
图4 用Structure 获得的平均可能性和ΔK 评估(利用Structure 软件12 次重复计算ΔK 的平均值)Fig.4 Plot of mean likelihood values (averaged across 12 runs) and estimate of ΔK for each possible value of K using the data obtained from Structure software

图5 克氏原螯虾9 个群体所有个体的遗传结构图Fig.5 Output from the program Structure assuming (K=3),(K=4) and (K=5) nine populations of P.clarkii
3 讨论
SSR 标记因具有多态性高、易操作及共显性遗传等特点,已被广泛应用于水产动物种质资源和选择育种等研究中[5-6]。本研究采用的10 对微卫星位点的平均等位基因数(Na) 15.70 个,平均有效等位基因数(Ne) 4.76 个,与Yue 等[12]、Zhong 等[23]和邢智珺等[14]的结果相比均较高,具有高度多态性,但仍低于曹玲亮等[24]获得的平均等位基因数,这与检测技术和样本收集的差异密切相关,即便使用相同的微卫星位点,但在采用不同检测技术进行分析时仍有可能表现出较大差异,通常认为先进的检测技术是等位基因检测的分辨率和准确性的有效保障[25]。多态性信息含量(PIC)为0.31~0.92,根据相关标准[26],高度多态性位点的PIC大于0.50,而小于0.25 的为低度多态性位点,本研究中除了PCL28 位点为中度多态性外,其他均属于高度多态性位点,均能用于各群体的遗传多样性分析。
克氏原螯虾作为一种入侵物种,由于其具有较高的商业价值,已成为中国最重要的淡水水产资源之一[26]。研究者们通过采集我国不同地区种质的克氏原螯虾群体来研究我国克氏原螯虾的入侵路线,发现我国的克氏原螯虾已具有较高的遗传多样性[12-14,24]。为了满足消费者的需求,我国各地区开展了大量的克氏原螯虾的人工饲养,由于不同的生长条件和管理差异,从而导致了不同种群之间不同的质量水平和遗传多样性。本研究中,9 个群体均具有较高的遗传多样性,其中,XC 群体的遗传多样性显著高于大多数群体(P>0.05),可能与其受开发利用程度较低有关。而JL 群体的遗传多样性均显著低于大多数群体,应与多代人工选择造成较高种质纯度有关,2 个人工养殖群体所表现出的遗传多样性水平差异,可能与其育种技术及育种的基础群体相关。总体上,安徽人工养殖群体的遗传多样性均高于对照群体,与Zhong 等[23]和李艳和[13]报道的群体相比均保持了相对较高的遗传多样性。这可能是由于从各种种群中多次引入不同的个体和不同的养殖条件,产生了迅速的遗传变异,使得种群具有较高的遗传多样性。Hardy-Weinberg 平衡表明,9 个群体均存在显著的杂合子缺失,其中HF 群体表现更突出,类似于Yue 等[12]的发现,这可能是由于创始人效应和非随机交配引起[27-28],安徽作为我国克氏原螯虾主要产区之一,其养殖群体应该存在大量的引种现象。由于克氏原螯虾的繁殖能力较强,在人工养殖过程中极易造成亲缘相近的克氏原螯虾个体间发生交配,因此,有必要加强不同区域间的群体进行交配,以改善近亲繁殖的现状。
遗传结构是判断新物种适应新栖息地能力的主要因素[29]。根据AMOVA 结果显示,变异主要发生在种群内(88.58%),只有很小一部分发生在种群间,与邢智珺等[14]和Yi 等[30]的研究结果一致,人工养殖种群与野生种群的AMOVA 结果相同。大多数学者认为,不同群体具有不同的遗传分化[30-31],物种间的遗传分化与基因交流密切相关[32]。本研究中,在安徽地区的人工养殖群体中,WH 与JH 群体间基因流水平最高(Nm=9.710),遗传分化指数最小(Fst=0.025),说明2 群体间的基因交流较为广泛(Nm>4),2 个群体间存在相互引种的可能。HF 与TL 群体间基因流水平最低(Nm=1.028),遗传分化指数最高(Fst=0.196),说明2 群体间基因交流较为匮乏,群体间存在显著遗传分化(Fst>0.15),这可能是由于长期封闭养殖模式和人工引入造成的,因为本研究WH 和HF 群体是人工养殖群体,野生群体不能进入与之交配,造成类似自然地理隔离,降低了发生基因交流的可能性。因此,可以通过改善育种技术和降低捕捞强度来加强群体间的基因交流。
由于人类传播、遗传漂变和种群规模等多种因素的影响[12],使得遗传距离与地理距离相关性显著降低。本研究中的JL 群体在地理上均远离其他8 个群体,但克氏原螯虾具有较高的商业价值,因此他们之间的遗传距离不同可能是引种程度不同所造成的。9 个克氏原螯虾群体基于Nei 氏遗传距离构建的UPGMA 系统树显示,9 个群体可分为5 组,即JH、WU、GH 和MAS 群体聚为一组,XC 和HF 群体同属于一组,而TL、XH 和JL 群体分别自成一组,与Structure 基于个体遗传组成的群体模拟分析的研究结果相类似。结合对群体遗传聚类及遗传结构等分析可以发现,XC和HF 群体的大多数个体被分配到相同的遗传群体中,这可能与它们引进了相同的基础选育群体有关。而WH 与MAS 群体、JH 群体、GH 群体及XH 群体的一些个体相较混杂,极有可能是它们之间相互引种所导致的。在野生群体中,TL 群体的个体遗传结构相对单一,与其他群体的个体有着明显的区别,说明TL 群体的开发利用程度较低。相对自然选择,位点等位基因频率与人工干预程度有着重要的影响,而且位点等位基因频率改变和群体遗传结构纯化与人工选择过程的相关性更明显。
总之,通过对这9 个群体的遗传多样性和遗传机构分析,可以看出安徽地区的人工养殖群体具有较高的遗传多样性,其中XC 群体的遗传多样性最高,遗传结构也较为合理,可以作为人工引种的基础选育群体。
(作者声明本文无实际或潜在的利益冲突)
猜你喜欢
特产研究(2022年6期)2023-01-17
世界科学技术-中医药现代化(2022年3期)2022-08-22
四川动物(2017年4期)2017-07-31
西南农业学报(2016年6期)2016-04-16
法医学杂志(2015年4期)2016-01-06
实用手外科杂志(2015年4期)2015-08-27
实用手外科杂志(2015年3期)2015-08-27
实用手外科杂志(2015年3期)2015-08-27
集美大学学报(自然科学版)(2015年4期)2015-02-28
河北遥感(2014年3期)2014-07-10
