
IL-33在哮喘合并间歇性缺氧发病过程中的作用
2023-11-02 00:52姚巧玲宋海辰于文燕马小娟
新疆医科大学学报 2023年10期
张 艳, 姚巧玲, 宋海辰, 于文燕, 孙 湛, 马小娟
(1新疆医科大学基础医学院病理生理学教研室, 2新疆地方病分子生物学重点实验室,3新疆医科大学基础医学院生理学教研室, 乌鲁木齐 830017; 4新疆医科大学第一附属医院儿内一科, 乌鲁木齐 830054)
哮喘(Asthma)是一种以可逆性气流受限、气道高反应性和气道炎症为特征的异质性疾病。阻塞性睡眠呼吸暂停综合征(Obstructive sleep apnea syndrome,OSAS)以间歇性缺氧为标志,上呼吸道完全阻塞(呼吸暂停)或部分阻塞(低通气)反复发作是其主要特征。OSAS与心血管疾病、胰岛素抵抗、神经损伤和死亡率增加有关[1]。哮喘和OSAS有相似的合并症和潜在的病理生理学重叠。
白介素-33(Interleukin-33,IL-33)属于白介素-1(Interleukin-1,IL-1)家族成员,是最新发现的一种与炎性疾病相关的促炎细胞因子,在调节机体血管生成以及炎症发生过程中具有重要作用[2]。Cayrol等[3]研究发现,IL-33与哮喘的关系密切,能够参与调节多种与哮喘相关的细胞及细胞因子。Sozer等[4]研究发现OSAS组患者血浆IL-33表达明显高于对照组。Perez等[5]指出IL-33可以作为一种前炎性细胞因子,活化炎症细胞产生多种炎症因子。故IL-33作为警报蛋白间接增加IL-1、IL-6、C反应蛋白等炎症因子的表达,促进了慢性炎症性疾病的发展。因此认为,慢性炎症状态会增加组织 IL-33 的含量,而 IL-33 可能与持续的炎症信号协同作用,从而进一步促进炎症和组织损伤。此外,有报道表明IL-33作为促进血管生成的因子,可促进皮肤肥大细胞和人角质形成细胞系HaCaT产生血管内皮生长因子(Vascular endothelial growth factor,VEGF)[6],而VEGF在血管生成过程中发挥着不可替代的作用,尤其是对于胚胎发育、伤口愈合和肿瘤生长过程中的新生血管生成[7]。
目前关于哮喘合并OSAS相互关联的机制研究较少,且关于二者之间发病机制以及病理生理过程存在一定的争议。本研究用C57BL/6J小鼠建立哮喘合并间歇性缺氧模型,通过研究小鼠肺组织IL-33、VEGF以及代表性炎症因子(IL-6、TNF-α)等指标的变化,探讨IL-33在哮喘合并间歇性缺氧发病过程中的作用。
1 材料与方法
1.1 动物选取健康雌性C57BL/6J小鼠32只,6~8周龄,体重18~20 g。以随机数字表法将小鼠分为对照组(NS-RA组)、间歇性缺氧组(NS-IH组)、哮喘组(OVA-RA组)和哮喘合并间歇性缺氧组(OVA-IH组),每组各8只。小鼠由湖南省斯莱克景达有限公司提供,许可证号:SCXK(湘)2019-0004,本研究严格按照国际实验动物护理评估与认证协会指南中的建议进行。
1.2 研究方法
1.2.1 造模 (1)哮喘模型建立:将OVA-RA组和OVA-IH组小鼠分别于第1天和第15天腹腔注射0.2 mL/只致敏液,致敏液由0.1mg卵清蛋白(OVA)+1 mg氢氧化铝配制而成;第22天雾化吸入1% OVA溶液,每次30 min,每周3次,连续8周。(2)间歇性缺氧模型建立:在第28天将NS-IH组和OVA-IH组小鼠暴露于间歇性缺氧(intermittent hypoxia,IH)环境下。暴露于IH是通过气体控制输送系统进行的[8],在IH控制舱内每分钟氧浓度在7%~21%呈正弦函数波动,每日暴露12 h(9∶00~21∶00),连续4周。(3)NS-RA组小鼠以生理盐水代替OVA处理,在相同的舱内以常氧(Room air,RA)条件代替间歇性缺氧处理。
1.2.2 Quantitative Real-time PCR检测IL-33和VEGF mRNA的表达 采用经典的Trizol法提取小鼠肺脏组织的总RNA,所用试剂及耗材均为RNase-Free型。IL-33引物序列: 上游引物, 5′-AATGCTTCACGGTGGAATAG-3′;下游引物,5′-GAAAGAAAGCTGAGGGAGTAG-3′。VEGF引物序列:上游引物,5′-TGTACCTCCACCATGCCAAGT-3′;下游引物,5′-CGCTGGTAGACGTCCATGAA-3′。β-actin引物序列:上游引物,5′-GTGACGTTGACATCCGTAAAGA-3′;下游引物,5′-GCCGGACTCATCGTACTCC-3′。反应条件:94℃ 30 s;94℃ 5 s,60℃ 30 s,共40个循环。按照制造商的说明书,使用TransScript®one-Step Gdna Removal and CDNA Synthesis SuperMIX试剂盒(AT311-03;TRAN全式金,中国北京)进行逆转录反应。使用PerfectStartTMGreen QPCR SuperMIX试剂盒(AQ601-02;TRAN全式金,中国北京)在实时荧光定量PCR反应系统(Life Technologies,USA)中进行实时荧光定量分析,反应结束后分析熔解曲线,并将数据归一化处理,采用2-ΔΔCt对基因表达水平进行定量分析。
1.2.3 Western Blot检测IL-33和VEGF蛋白的表达 取40 mg肺组织,加入蛋白裂解液(Thermo,USA)后用研磨器研磨,冰上裂解离心并吸取上清。然后用BCA法进行蛋白定量。随后通过12%SDS-PAGE凝胶分离40 μg蛋白样品并转移至PVDF膜,室温下用5%BSA封闭2 h,然后在4℃下分别与IL-33(1∶3 000,兔抗,Affinity)、VEGF(1∶1 000,兔抗,proteintech)、β-actin(1∶2 000,兔抗,proteintech)一抗孵育过夜。接下来,用相应二抗(1∶5 000,HRP偶联山羊抗兔IgG;proteintech,USA)在室温条件下孵育1 h,并使用增强的化学发光试剂盒(biosharp,中国)进行显影。采用Image J进行统计分析。
1.2.4 酶联免疫吸附(ELISA)法检测肺组织IL-33、IL-6以及TNF-α的变化 将肺组织于冰上充分研磨后,以5 000 g离心10 min后取上清液进行检测。使用ELISA试剂盒检测肺组织中IL-33、IL-6以及TNF-α的浓度。
1.2.5 肺组织病理学分析 将小鼠肺脏上叶浸入10%福尔马林固定12 h,修整组织块,常规石蜡包埋切片,进行苏木素-伊红(HE)染色。将石蜡切片常规脱蜡至水,并按照试剂盒说明书中的方法进行Masson染色,普通光学显微镜来捕获图像,并采用Image定量分析染色结果。
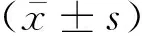
2 结果
2.1 各组小鼠肺组织IL-33和VEGF mRNA表达变化与NS-RA组相比,其余3组小鼠肺组织中IL-33与VEGF mRNA表达均增加,其中OVA-IH组增加最显著,差异有统计学意义(P<0.001),见表1、图1。

注:A,IL-33 mRNA相对表达量;B,VEGF mRNA相对表达量;与NS-RA组相比, ###P<0.001。图1 各组小鼠肺组织中IL-33和VEGF mRNA的表达

表1 各组小鼠肺组织中IL-33和VEGF mRNA的表达
2.2 各组小鼠肺组织中IL-33和VEGF蛋白表达变化与NS-RA组相比,OVA-IH组小鼠肺组织中IL-33与VEGF的表达明显增加(P<0.01~0.05);与NS-IH组和OVA-RA组相比,OVA-IH组小鼠肺组织中IL-33与VEGF的表达有所增加,但差异无统计学意义(P>0.05),见表2、图2。

注: A, 各组蛋白免疫印迹图; B-C, 各组蛋白相对表达水平; 与NS-RA组相比, #P<0.05, ##P<0.01。图2 各组小鼠肺组织中IL-33和VEGF的蛋白表达
表2 各组小鼠肺组织中IL-33和VEGF蛋白的表达
2.3 各组小鼠肺组织中炎症因子水平变化与NS-RA组相比,其余3组小鼠肺组织中IL-33和IL-6水平升高,其中OVA-IH组升高最明显,差异有统计学意义(P<0.01~0.05);与NS-RA组相比,OVA-IH组小鼠肺组织中TNF-α水平升高,但差异无统计学意义(P>0.05);与NS-IH组相比,OVA-IH组小鼠肺组织中TNF-α水平升高且差异有统计学意义(P<0.05),见表3、图3。

注: A-C, 各组小鼠肺组织炎症因子水平; 与NS-RA组相比, #P<0.05, ##P<0.01; 与NS-IH组相比, *P<0.05。图3 各组小鼠肺组织中炎症因子的水平变化

表3 各组小鼠肺组织中IL-33、TNF-α和IL-6的水平变化
2.4 IL-33与TNF-α、IL-6水平的相关性Pearson相关性分析显示,小鼠肺组织中IL-33和TNF-α(r=0.576 0,P<0.01)、IL-33和IL-6(r=0.765 3,P<0.000 1)均呈中度正相关,见图4。
2.5 肺组织病理变化各组小鼠肺组织HE染色后镜下观察可见,NS-RA组肺组织支气管结构光滑且完整,细胞排列整齐,管腔及其周围未见明显异常;其余3组小鼠气道增厚,气道上皮细胞水肿,杯状细胞增殖,肺泡壁增厚,平滑肌增生,支气管变形,支气管壁结构受损,管腔内有脱落细胞,肺泡壁毛细血管充血和水肿增加,血管壁增厚,支气管以及血管周围有大量炎性细胞浸润和蓄积(图5A)。与NS-RA组相比,其余3组气道黏膜中的胶原纤维表达增加,并有明显的蓝色胶原沉积(图5B、图6)。对Masson染色阳性区域进行定量分析并测量气道胶原沉积指数发现,与NS-RA组(7.11±0.98)%相比,NS-IH组(14.13±1.59)%、OVA-RA组(18.31±1.49)%和OVA-IH组(19.73±1.28)%的气道胶原沉积指数均升高,OVA-IH组升高最为明显(P<0.001)。

注: 与NS-RA组相比, #P<0.05, ###P<0.001。图6 各组小鼠肺组织Masson染色阳性面积比较
3 讨论
近年来,越来越多的证据表明OSAS与肺部疾病(慢性阻塞性肺病、哮喘、间质性肺疾病和肺动脉高压)之间存在双向关系,即每种疾病都会对另一种疾病产生有害影响。相关研究报道显示,哮喘合并OSAS的发病机制可能与神经机械反射性支气管收缩、局部和全身炎症、OSAS诱发的心功能不全对呼吸困难的间接影响、血管生成和瘦素相关气道改变等病理生理过程有关[9]。采用卵清蛋白诱导激发建立哮喘小鼠模型以及间歇性缺氧建立间歇性缺氧小鼠模型已在许多研究中被广泛应用[10-11]。本研究在此模型基础上观察哮喘合并间歇性缺氧小鼠肺组织中IL-33、VEGF以及炎症因子白介素-6(IL-6)、肿瘤坏死因子-α(TNF-α)的表达。
以往的研究发现,IL-33的mRNA表达于人和鼠的多种器官和细胞。在蛋白水平,IL-33主要表达在成纤维细胞、上皮细胞和内皮细胞[12]。本研究结果显示,与对照组相比,各处理组小鼠肺组织中IL-33 mRNA和蛋白水平显著增加。气道炎症是哮喘最常见的病理改变[13]。越来越多的研究也表明OSAS为低度慢性炎症性疾病[14]。炎症发生时炎症因子表达增加,IL-33作为炎症因子还参与肥大细胞、嗜碱性粒细胞、嗜酸性粒细胞和自然杀伤细胞的活化[15]。有研究表明,哮喘[16]与OSAS[17]患者血清中IL-33均增加,这与本研究的结果一致。
炎症发生时,抗炎与促炎的失衡导致机体分泌大量促炎和抗炎因子。IL-33的靶标包括参与Ⅰ型和Ⅱ型免疫和调节反应的免疫细胞,可参与Th2细胞的成熟,进而促进其他炎症因子释放增加[18]。本研究对炎症因子IL-6和TNF-α的定量检测发现,哮喘合并间歇性缺氧时IL-6和TNF-α的水平升高,提示间歇性缺氧可能使哮喘小鼠炎症反应加重,而NS-IH组小鼠肺组织中TNF-α水平与NS-RA组相比略微降低,Corral等[19]也发现在阻塞性睡眠呼吸暂停患者牙龈沟液中的TNF-α水平显著低于健康人群,推测可能是间歇性缺氧时,TNF-α作为炎症介质具有抗炎和促炎双重功能。相关性分析表明小鼠肺组织中IL-33和TNF-α、IL-33和IL-6均呈中度正相关,进一步提示IL-33可能与持续的炎症信号协同作用,使炎症反应放大进一步加剧组织损伤。
炎症反应又与血管生成息息相关,VEGF在其中起着至关重要的作用。慢性炎症可导致血管生成,血管生成是多种非肿瘤性慢性炎症的重要组成部分[20]。在肺血管发育过程中的各种细胞因子中,VEGF是肺血管发育所需的最重要、最有效、最直接、最专一的生长因子,此外VEGF还可刺激血管内皮细胞的有丝分裂和血管的发生,提高单层内皮的通透性[21]。研究表明,在炎症性肠病小鼠肠黏膜中VEGF显著上调[22]。除了诱导炎症反应外,IL-6和TNF-α也可能是生理和病理血管生成的重要调节分子,研究认为IL-6可能通过激活Janus激酶/信号传导蛋白和转录激活物(JAK/STAT3)信号通路增加巨噬细胞中VEGF的表达[23],而TNF-α可能通过活性氧依赖性激活β-catenin从而上调RPE细胞中VEGF表达[24]。
本研究结果显示,与对照组相比,各处理组小鼠肺组织中VEGF mRNA和蛋白水平明显增加。在正常肺组织中,由肺泡上皮衍生的各类细胞均表达VEGF,尤其是肺泡Ⅱ型细胞以及细支气管clara细胞、细支气管周围小动脉的平滑肌细胞[25]。气道重塑是哮喘的关键特征,其中血管生成对于气道重塑尤为重要[13]。间歇性缺氧是OSAS的主要特征,而氧供与氧耗失衡可引起血管生成[26]。VEGF是哮喘发病过程中的一个非常重要的分子生物学因素,支气管哮喘患者痰液中VEGF的表达与哮喘严重程度和气道炎症密切相关[27]。OSAS可引起严重的低氧血症及睡眠紊乱,低氧-复氧的交替过程可诱导机体缺氧诱导因子-1α(HIF-1α)高表达从而导致VEGF 的表达增加[28]。本实验通过形态学观察也发现,OVA-IH组小鼠肺组织炎症反应加重、气道结构改变、气道与周围血管胶原沉积均增加。
综上所述,本研究揭示了哮喘合并间歇性缺氧的发病过程中,IL-33与炎症因子协同作用导致VEGF表达增加可能参与了其发病过程,这可能为临床治疗提供一个新的思路,但IL-33是否是介导哮喘合并间歇性缺氧疾病时血管生成相关气道改变的关键因子还有待进一步的研究。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
保健医苑(2022年1期)2022-08-30
南京林业大学学报(自然科学版)(2021年5期)2021-10-13
中老年保健(2021年6期)2021-08-24
国际呼吸杂志(2019年22期)2019-12-09
国际呼吸杂志(2019年5期)2019-03-30
国际呼吸杂志(2019年3期)2019-03-01
国际呼吸杂志(2019年2期)2019-02-14
广西林业科学(2016年3期)2016-03-16
