
铁基催化剂活性相调控及其催化CO2加氢制线性α-烯烃研究进展
2023-11-01 08:18高新华夏世钦江永军郭庆杰
天然气化工—C1化学与化工 2023年5期
高新华,夏世钦,梁 洁,江永军,张 伟,郭庆杰,王 晨,段 斌
(1.宁夏大学 化学化工学院 省部共建煤炭高效利用与绿色化工国家重点实验室,宁夏 银川 750021;2.国家能源集团宁夏煤业有限责任公司煤炭化学工业技术研究院,宁夏 银川 750411;3.国家煤及煤化工产品质量检验检测中心(宁夏) 宁夏计量质量检验检测研究院,宁夏 银川 750200)
化石能源是目前主要的能源形式,在我国能源结构中占主导地位[1]。人们对化石能源的过度依赖使大气中二氧化碳(CO2)浓度增加,导致全球气候变化异常,因此降低大气中的CO2浓度已成为一项必要的任务[2]。根据国际原子能机构发布的《2022年二氧化碳排放报告》,截至2022 年中国CO2排放总量超过114.8 × 108t,排放总量居世界首位。为应对这一环境问题,中国已经率先承诺在2030年前后实现“碳达峰”。降低大气中CO2浓度主要有以下3种方式:(1)采用清洁能源代替传统化石能源;(2)CO2的捕集及封存;(3)CO2转化利用[3]。以CO2作为碳源,通过CO2催化加氢反应将CO2与“绿氢”转化为一系列燃料和高附加值化学品,既可以实现煤炭资源清洁高效利用,又可以缓解温室效应,如图1 所示[4-5]。近年来,科学家在CO2加氢制取甲烷、甲醇和低碳烯烃的催化剂设计方面已经取得了较大突破[6-7]。
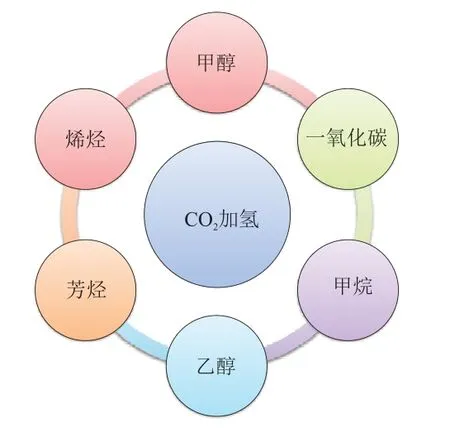
图1 CO2加氢产物Fig.1 CO2 hydrogenation products
线性α-烯烃(LAOs)是碳链末端含有一个C==C键且碳数不小于4的直链烯烃,是生产润滑油、洗涤剂和聚烯烃的重要原料[8-9]。通常C4~C10LAOs 可以作为工业生产增塑剂的原料,C10~C20LAOs 可以用于生产洗涤剂、润滑油等化学品[10]。目前,工业上主要通过石蜡裂解[11]和低碳烯烃齐聚[12]等石油化工路线制取LAOs。由于石油化工路线的不可持续性,通过CO2加氢反应制取LAOs 具有重要意义。目前,CO2加氢制LAOs主要经由费托合成(FTS)路径(CO2-FTS 路径)实现,该路径主要包含两个过程:CO2和氢气(H2)经过逆水煤气变换(RWGS)反应生成一氧化碳(CO);CO 发生加氢反应生成LAOs,分别如式(1)和式(2)所示。Fe基催化剂兼具RWGS和FTS 反应活性,是CO2加氢制烯烃反应中应用最广泛的催化剂[13-15]。
Fe 基催化剂在活化和反应过程中会经历复杂的物相变化,一般认为四氧化三铁(Fe3O4)和碳化铁(FexCy)分别是RWGS和FTS反应的活性相,特别是χ-Fe5C2对链增长反应起关键作用[16]。因此,提高Fe 基催化剂中χ-Fe5C2相的含量有利于C—C 偶联反应生成LAOs。反应过程中生成的H2O 会使χ-Fe5C2被氧化为Fe3O4,导致催化剂失活、LAOs 选择性降低,所以维持χ-Fe5C2相的稳定是非常有必要的[17-19]。目前关于CO2加氢反应制取LAOs Fe 基催化剂活性相调控的研究报道较少。本文对CO2加氢制LAOs 的反应机理以及Fe 基催化剂物相演变过程、失活机制进行综述,对Fe 基催化剂活性相调控及其催化CO2加氢制LAOs 的研究进展进行总结,主要包括载体、助剂以及表面亲疏水性改性对Fe基催化剂活性相生成、稳定的影响,提出在该领域催化剂设计方面面临的挑战,以期为CO2高效转化利用提供借鉴。
1 CO2加氢制LAOs反应机理
明确CO2加氢制LAOs 反应机理有助于优化催化剂设计[20]。CO2加氢制烯烃主要分为以下两种路线:(1)甲醇中间体路线(CO2-MeOH)。CO2首先在金属氧化物上转化为甲醇,甲醇在分子筛酸位上合成烯烃,其反应产物以低碳烯烃为主[21-22]。(2)CO2-FTS路线。第一步RWGS反应(图2),CO2首先吸附在含有氧空位的Fe3O4结构上形成Fe(CO3)中间体,由于Fe(CO3)中Fe—O 的相互作用强于C—O的相互作用,因而Fe(CO3)会断键脱除CO,剩余的O—Fe—O结构会与H2反应脱水再次生成含有氧空位的Fe—O结构,从而实现RWGS循环反应。
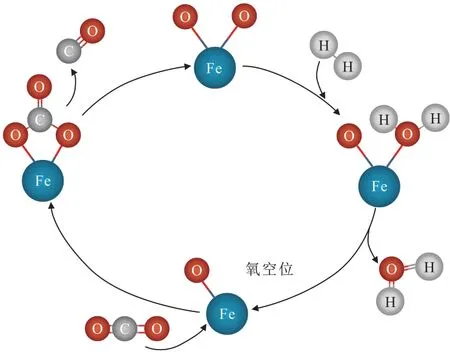
图2 RWGS反应机理[23]Fig.2 RWGS reaction mechanism[23]
第二步FTS 反应(图3),FexCy首先与H2反应生成烷烃,烷烃产物脱水形成含有碳空穴的FexCy结构。接着,此前RWGS 反应过程中生成的CO 会占据FexCy结构中的碳空穴位点形成Fe—C—O 结构,该结构中的O 会与H2反应脱水再次生成FexCy,从而实现CO 加氢制烃的循环反应过程。通常认为Fe3O4和χ-Fe5C2分别是RWGS反应和FTS反应的活性相,特别是χ-Fe5C2对链生长和LAOs 选择性起着关键作用[23-24]。因此,一般是在Fe 基催化剂上经由CO-FTS路线CO2加氢制取LAOs,通过调控Fe3O4与χ-Fe5C2的物质的量之比可以获得高LAOs选择性。
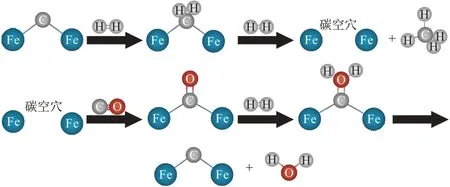
图3 FTS反应机理[23]Fig.3 FTS reaction mechanism[23]
2 Fe基催化剂失活机制
研究人员对Fe 基催化剂失活的主要原因有很多争论,主要有4种失活机制,分别为相变、积炭、烧结和中毒,如图4 所示[25]。在大多数工业气体原料中,硫化物(H2S、CH3SH、C2H5SH和COS等)可使Fe基催化剂迅速中毒失活[26]。
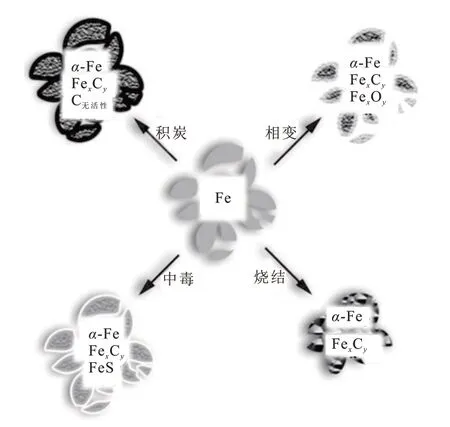
图4 Fe基催化剂的失活机制[25]Fig.4 Deactivation mechanisms of Fe-based catalysts[25]
2.1 相变
FexOy催化CO2加氢制烯烃可分为5个不同的动力学阶段[27]:第一阶段,反应物吸附在催化剂表面并碳化;第二、三阶段,RWGS反应产物在正在进行的碳沉积过程中占主导;第四阶段,FTS反应速率提高至稳态;第五阶段,维持这种稳态。在反应之前,催化剂的Fe相主要是α-Fe、Fe3O4和少量Fe2O3,随着反应的进行,Fe 和CO 解离产生的C 发生反应,形成FTS反应的活性相FexCy。H2O是FTS反应的主要反应产物之一,反应过程中H2O 的分压越高,反应速率越低。随着反应进行,会产生越来越多的H2O,将有活性的χ-Fe5C2相氧化为不具有FTS 反应活性的FexOy相,催化剂开始失活[28]。
2.2 积炭
在典型的FTS 反应过程中,催化剂表面会沉积非活性含碳化合物,例如石墨碳、无定形碳和焦炭,限制了反应气与活性相之间的接触,并污染、堵塞催化剂的表面活性位点,导致催化剂失活[29-31]。高分子量的蜡会填充到催化剂孔内,从而减缓反应物的扩散速率,而不溶性碳沉积物则会堵塞活性相,使催化活性降低,CH4选择性提高。积炭量取决于催化剂的预处理条件,与完全渗碳的催化剂相比,预还原催化剂更容易发生碳污染[32-33]。
2.3 烧结
所有Fe 基催化剂都有一定程度的活性相烧结失活现象。一般来说,烧结可描述为小晶粒由于成熟或迁移和聚结现象而生长的过程。烧结会因多种原因对催化性能产生负面影响,由于较小的催化剂晶体会因烧结而凝聚成较大的晶体,导致孔结构损失、催化剂结构坍塌,活性相转化为非活性相[34-35]。
3 Fe基催化剂活性相调控及其催化CO2加氢制LAOs研究进展
研究人员通过加入助剂、载体和表面改性等方法以促进χ-Fe5C2活性相的生成、调控Fe3C4/χ-Fe5C2比例(物质的量之比,下同)、维持χ-Fe5C2的稳定,如表1所示[36]。
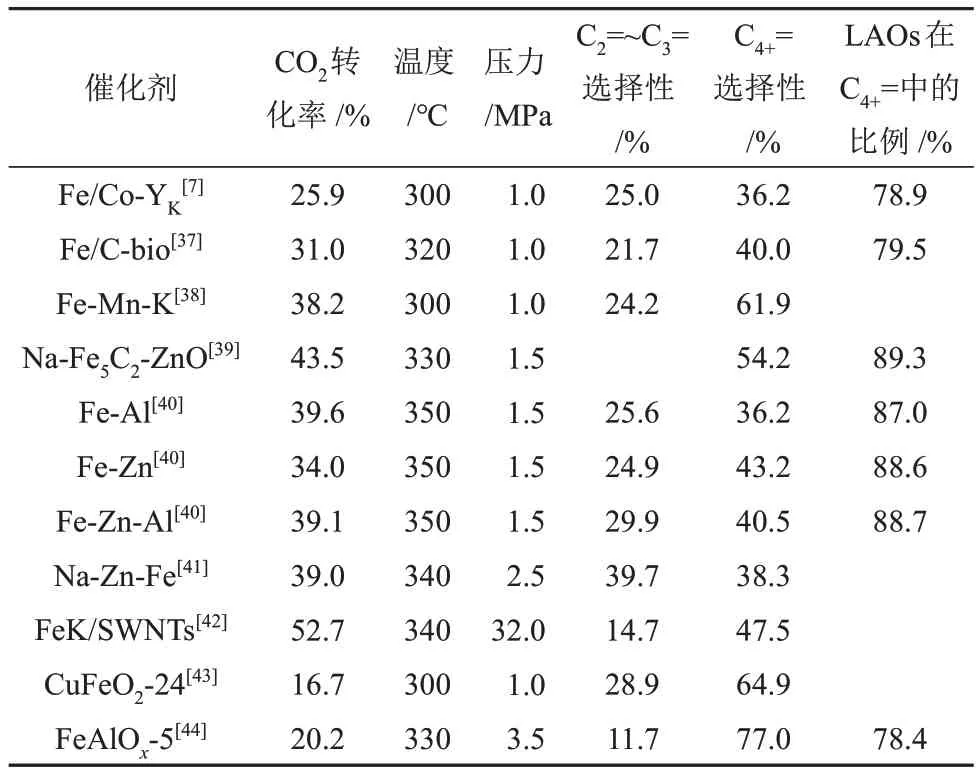
表1 经CO2-FTS路线制LAOs 中Fe基催化剂的对比Table 1 Comparison of Fe-based catalysts in CO2 hydrogenation of LAOs by CO2-FTS route
3.1 促进χ-Fe5C2相生成
研究发现,在Fe 基催化剂中加入碳材料载体,Na、K 等助剂,有助于活性金属的活化、碳化,促进χ-Fe5C2活性相的生成[45]。
3.1.1 碳基载体促进χ-Fe5C2相生成
碳材料具有较高的稳定性、耐水性、渗碳能力和适度的金属-载体相互作用,可促进活性FexCy的形成,是Fe基催化剂的理想载体之一[46-47]。WANG等[42]开发了单壁碳纳米管(SWNTs)负载的FeK 催化剂(FeK/SWNTs),在高空速(9000 mL/(g·h))下,CO2转化率为52.7%,C4+=选择性达47.5%,CO选择性仅为9.6%。这种优异的性能归因于SWNTs具有大曲率的高倾斜度来贡献电子,促进CO*的解离、χ-Fe5C2的形成,提高催化剂表面吸附的CO2和H2物质的量之比(记作“C/H”,下同)。此外,他们还发现FeK/SWNTs催化剂对低碳烯烃的吸附强度较高,这有利于低碳烯烃进一步转化为高碳烃。MALHⅠ[48]制备了多种多壁碳纳米管(MWNTs)负载铁基催化剂(FeZnNa/MWNTs、FeZnK/MWNTs 和FeZnRb/MWNTs)。结果表明,MWNTs 的加入显著影响Fe 基催化剂的形态、还原、物相结构和吸附行为,进而影响了CO2加氢制LAOs反应活性。具体而言:(1)加速了χ-Fe5C2活性相的形成;(2)富电子MWNTs 表面具有给电子能力,在FTS过程中促进C—O键断裂并增强Fe—C键,加速了碳单体的合成,提高了催化剂表面的C/H;(3)对CO和低碳烯烃的结合强度更高,有利于生成LAOs。FeZnNa、FeZnK和FeZnRb催化剂的高碳烯烃(C5=~C8=)选择性分别从27.1%、27.0%和24.2%提高到32.2%、31.1%和28.6%,CH4和CO 副产选择性均降低。此外,催化剂在运行100 h 后仍表现出优异的催化性能。
3.1.2 碱金属助剂促进Fe5C2相生成
Na 和K 助剂可作为电子供体,调节Fe 位点的电子性质,提高催化剂的表面碱度,有效降低CO2解离的能垒,促进χ-Fe5C2的形成[49-50]。RAMⅠREZ等[51]报道了Fe2O3@K2CO3催化剂用于CO2加氢反应,反应过程中K与CO2在催化剂表面反应形成了3种K相(K2CO3、KHCO3和KOOCH),并在反应条件下相互转化。CO2与K2CO3反应生成KCOOH 和CO,生成的CO 进行FTS 反应合成烯烃。另外,在该研究中还发现K 助剂可以诱导Fe/C 催化剂形成了更活跃的Fe5C2-K2CO3界面,促进了K 和Fe 之间的电子转移,提高了LAOs 选择性。CO2转化率为32.6%时,C4=~C20=选择性达56.6%。此外,Fe/C 催化剂与K助剂之间适当的接触距离有利于C—C 偶联反应,抑制CH4副产的生成。一般情况下,少量Na助剂就足以提高Fe 基催化剂反应活性和LAOs 选择性。ZHANG 等[41]研究了Na 助剂的作用:促进CO2吸附和高活性Fe5C2的形成,从而提高了CO2的转化率。此外,Na 的加入可以减弱烯烃在Fe 催化剂表面的吸附,从而抑制烯烃的二次加氢反应。此外,Na 的加入使Fe5C2的粒径缩小,增加Fe5C2表面的Na 含量,可以抑制Fe5C2相的还原,有利于烯烃的生成。Na 助剂改性的Fe 基催化剂上CO2转化率由18%提高至31%,LAOs 选择性由1.5%提高至44.0%,CH4副产选择性由48%显著降低至12%[52]。
3.1.3 其他助剂促进Fe5C2相生成
Cu 助剂常被用于提高Fe 基催化剂CO2加氢活性,反应中Cu 氧化物首先被H2还原形成Cu0,随后与FeOx物种结合,反应过程中催化剂表现出较高的RWGS反应活性。这是因为Cu0为H2解离提供了活性中心,通过氢溢出效应促进表面氧物种的还原。此外,反应过程中较活跃的表面氧物种与FeOx-Cu0界面存在的表面羟基密切相关,这直接影响了CO2活化速率[53]。除Cu0外,各种前驱体中Cu 的其他氧化态也可用于CO2在Fe 基催化剂上加氢制烯烃。CHOⅠ等[43]发现,与尖晶石CuFe2O4和Cu2O-Fe2O3前驱体相比,以铜铁矿CuFeO2作为前驱体获得的Cu-Fe催化剂表现出更高的烯烃/烷烃选择性之比(O/P= 7.3)和C5+烃选择性(66.3%)。这主要是因为CuFeO2中得Cu+能够显著促进Fe 的还原和渗碳,从而形成活性更强的χ-Fe5C2相。
除金属助剂外,从生物质中提取的含K、Si、Mg和Ca等多种元素的生物助剂,也可被用于修饰用于CO2加氢合成LAOs的Fe基催化剂。GUO等[37]以玉米芯灰为原料,采用生物助剂改性Fe 基催化剂,对C4=~C18=选择性为50.3%,其中LAOs占80%,CH4选择性降低到11.8%。与化学助剂相比,生物助剂具有优异的性能,这主要归功于生物助剂中多种元素的协同作用。生物助剂的存在促进了Fe 物种的渗碳,增加了Fe5C2含量[54]。
3.2 调控Fe3O4/χ-Fe5C2比例
一般认为Fe3O4和Fe5C2分别是RWGS和FTS反应的活性相,加入适配的载体、助剂可以合理调控Fe 基催化剂中Fe3O4/χ-Fe5C2比例,有利于提高CO2加氢制LAOs催化性能。
LⅠU 等[55]发现载体可以调控FexCy组成以及FexOy/FexCy的比例(物质的量之比),相较于SiO2、Al2O3、ZrO2和碳纳米管(CNT)等载体,ZrO2负载的催化剂中Fe5C2相含量最高。
ZHANG 等[56]将适量Zn 加入Fe 基催化剂用于催化CO2加氢反应,LAOs 的时空产率是纯Fe 基催化剂的2.4 倍,C4=~C20=选择性提高到60.7%,其中LAOs 占比为89.3%,CO2转化率达43.5%。该团队还揭示了Zn 在Fe5C2-ZnO 催化剂中的电子效应,如图5 所示。该报道认为还原后催化剂表面的Fe5C2与Zn0会同时暴露在CO2和H2的混合气氛中,CO2和体系中的H2O 会氧化Fe5C2和Zn0,催化剂表面形成高分散的FeOx和ZnO。在这个过程中,Zn0到ZnO的转变过程促进了CO2活化。FexOy催化CO2加氢生成了CO 和H2O,随后在未被氧化的Fe5C2上催化CO 加氢生成烃。随着高分散FexOy生成位点的增加,RWGS 反应活性提高,产生更多的CO,CO 在FexCy上吸附解离形成聚合碳单体,进一步发生链增长反应生成长链烃产物。前驱体中的FexOy可以被CO还原并碳化成FexCy,同时ZnO被还原为Zn0。因此,催化剂结构在反应过程中形成动态平衡。调控Zn浓度可控制催化剂表面的Fe3O4/χ-Fe5C2比例和Zn2+/Zn0比例(物质的量之比),使反应整体进入稳定阶段。作者认为ZnO-Zn/Fe5C2-FexOy之间的动态平衡决定了CO2加氢生成LAOs 的速率。这种界面活性位点的动态平衡结构为后续研究多功能催化剂的活性位点提供了很好的研究思路。
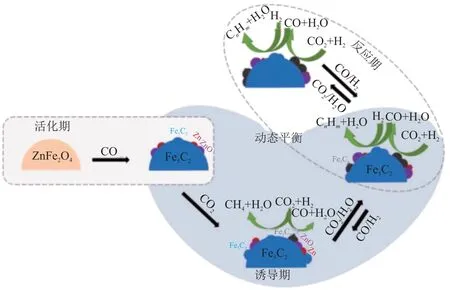
图5 Fe5C2-ZnO 催化剂在诱导和反应过程中表面结构的演变[56]Fig.5 Surface structure evolution of Fe5C2-ZnO catalysts during induction and reaction period[56]
碱土金属具有与碱金属相似的性质,但由于碱土金属熔点高,其结构更稳定。此外,碱土金属(Ca、Sr和Ba)通常具有ns2价层构型,可以将电子从s 轨道移动到d 轨道,是一种重要的助剂[57-58]。OREGE 等[57]报道了一种Sr 和Na 共修饰的Fe 催 化剂,该催化剂在合成高值烯烃(乙烯、丙烯和LAOs)方面表现出优异的活性,时空产率可达290 mg/(g·h),运行500 h 以上没有明显的失活,高值烯烃选择性达66.1%,CO2转化率为38%。这归因于Sr 和Na 在催化剂中不同位点(Fe5C2、Fe3C 和Fe3O4)上的协同作用。SrCO3促进了Fe 的分散、FexCy相的形成和稳定。同时,Sr有助于Na和Fe之间的电子相互作用。Sr 兼具结构和电子助剂的作用,促进了C—O 键的解离吸附和随后的C—C偶联,从而促进了CO2加氢反应。NaBaFe 催化剂中Ba 和Na 助剂共存加速了晶格耗氧,同时有利于产生更多的氧缺陷位点,增强了CO2的活化、Fe5C2的形成和中间物种的吸附。此外,Ba助剂可以抑制Fe5C2相的过度氧化,增强催化稳定性[52]。
3.3 维持Fe5C2相稳定
由于FexCy被氧化为FexOy是导致催化剂失活最关键的因素,因此研究人员通过表面改性、加入助剂等方法以提高FexCy相的稳定性[59-61]。
3.3.1 表面改性维持Fe5C2相稳定
在FTS 反应过程中,初级烯烃会发生二次反应导致其选择性降低,C1副产物选择性提高。研究发现,通过对催化剂进行表面亲疏水改性可以有效调变烯烃和H2O 的吸脱附行为,从而影响烯烃的二次反应,定向调控产物分布[59-60]。XU等[59]制备了一种疏水改性的FeMn@Si 催化剂,图6 为该催化剂的TEM照片。

图6 FeMn@Si催化剂的TEM照片[59]Fig.6 TEM images of FeMn@Si catalysts[59]
由图6 可以清晰看到FeMn@Si 为完整的核壳结构,SiO2壳是连接Fe 与疏水甲基基团的桥梁,有效抑制FexCy被H2O 氧化为FexOy,并缩短了H2O 在催化剂表面停留时间,抑制了水煤气变换(WGS)反应。CH4、CO2等副产选择性降低到22.5%,烯烃产率达36.6%。Mn 的加入促进了C—C 偶联反应,有利于生成长链烃。ZHANG等[60]在Fe5C2表面构建了一层疏水SiO2壳,发现随疏水壳含量增加,Fe5C2表面上H2O 分压增加、CO2加氢的活化能提高,导致CO2转化率降低。这是由于Fe5C2表面吸附的H2O提高了C—C偶联反应的能垒,使CH4的形成比其C—C偶联的竞争反应更容易进行,CH4选择性提高,C2+烃选择性降低。
3.3.2 助剂维持Fe5C2相稳定
在Fe基催化剂中加入ZnO不仅可提高Fe5C2含量,也有效抑制FexCy到FexOy的相变,维持催化稳定性。ZHANG 等[39]发现Zn 的引入显著抑制了Fe2O3的失活,这归因于在活化过程中Zn 向Fe2O3表面迁移,有效防止FexCy在反应中被H2O 和CO2氧化。ZHANG等[61]发现在FeZn催化剂中引入Na后,抑制了FexCy的氧化,从而提高了Fe5C2相的稳定性,进而提高了Fe 基催化剂CO2加氢制LAOs 选择性。Fe2Zn1 催化剂在运行200 h 后对C2=~C7=表现出高选择性,C4+=总选择性为81.9%,C4+=中LAOs 高达85.9%,CO2转化率为35.0%,而Fe2O3上CO2转化率由30.0%下降为12.8%。
综上,CO2加氢制LAOs 主要经由两步反应,Fe3O4和Fe5C2分别是RWGS反应和FTS反应活性中心。为获得高CO2转化率和LAOs 选择性,研究表明加入适配的助剂、载体以及通过表面改性可以促进Fe5C2生成、调控Fe3O4/χ-Fe5C2比例、维持Fe5C2相的稳定,有利于生成LAOs。但需要结合先进表征技术以揭示促进Fe5C2活性相生成、稳定的机制,以及反应中Fe物相演变过程[62-64]。
4 结语与展望
以CO2为原料,高选择性制取LAOs 不仅可以拓宽非石油路线生产LAOs 的途径,还可以助力碳减排,有效降低温室效应等气候问题。目前主要是通过CO2-FTS 反应路径将CO2转化为高碳烃。Fe3O4是RWGS 反应的活性中心,Fe5C2对链生长和LAOs 选择性起着关键作用。在设计催化剂时,通过改变载体,使金属-载体保持适中的相互作用,以及加入Na、K或Cu等助剂可以促进Fe物种渗碳,生成Fe5C2活性相。适配的载体、过渡金属或碱金属改性有利于维持不同界面位点的动态平衡,使Fe3O4/χ-Fe5C2处于适当比例。表面疏水改性可有效抑制FexCy被氧化,但催化剂表面疏水层不利于传质、导致CO2难以活化。目前来看,如何创新催化剂,实现CO2高效转化为LAOs选择性仍存在诸多瓶颈。
未来的研究重点应集中在以下几个方面:(1)利用不同助剂之间的协同作用合理选择载体,提高催化剂中的Fe5C2含量。(2)通过对Fe基催化剂表面疏水改性或加入适当助剂,抑制Fe5C2活性相的氧化。(3)加强对Fe5C2上CO2加氢制LAOs反应过程中链增长机理的研究,主要包括CO2活化、C—C 偶联和碳链终止反应的催化机理,为设计高效Fe基催化剂提供理论指导。
猜你喜欢
云南化工(2021年8期)2021-12-21
中国石油石化(2021年9期)2021-07-17
今日农业(2020年20期)2020-11-26
当代化工研究(2016年7期)2016-03-20
当代化工研究(2016年2期)2016-03-20
橡胶工业(2015年8期)2015-07-29
中国洗涤用品工业(2015年5期)2015-02-28
河南科技(2015年2期)2015-02-27
应用化工(2014年12期)2014-08-16
食品科学(2013年19期)2013-03-11
