
刺猎蝽属种间转录组比较分析
2023-10-30 12:11:20邓美菁
昆虫学报 2023年9期
邓美菁, 赵 萍
(1.南宁师范大学, 环境与生命科学学院, 南宁 530100; 2. 南宁师范大学, 地理与海洋研究院, 南宁 530100)
高通量测序技术获得的转录组数据及分析结果在昆虫生物化学、昆虫生理学、昆虫行为学及害虫防治等研究领域得到广泛应用(邹德玉, 2013; 李振, 2017; 代惠洁, 2018; 赵乐, 2018; 杨雪娇等, 2020; 覃英灿等, 2021),并且可以应用于相同物种的不同发育阶段或雌雄个体和不同组织的转录组数据比较(郑茜茜, 2017; 王亚冰, 2020; 赵星, 2021),为昆虫生长发育机制奠定理论基础。在生物分类和物种分化研究中,转录组数据也广泛应用于物种分化及其适应性研究领域(王胜, 2018; 叶航, 2020; 韩洁, 2022; 李珺, 2022)。
1 材料与方法
1.1 实验材料
齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽成虫样本分别采自贵州省雷山县、贵州省兴仁县和广西省龙州县(表1)。使用成虫活体作为实验材料,各地成虫出现时期不同,采集时间不同,采集活体实验材料在实验室以果蝇Drosophilasp.活虫作为食物饲养,统一送公司测序。
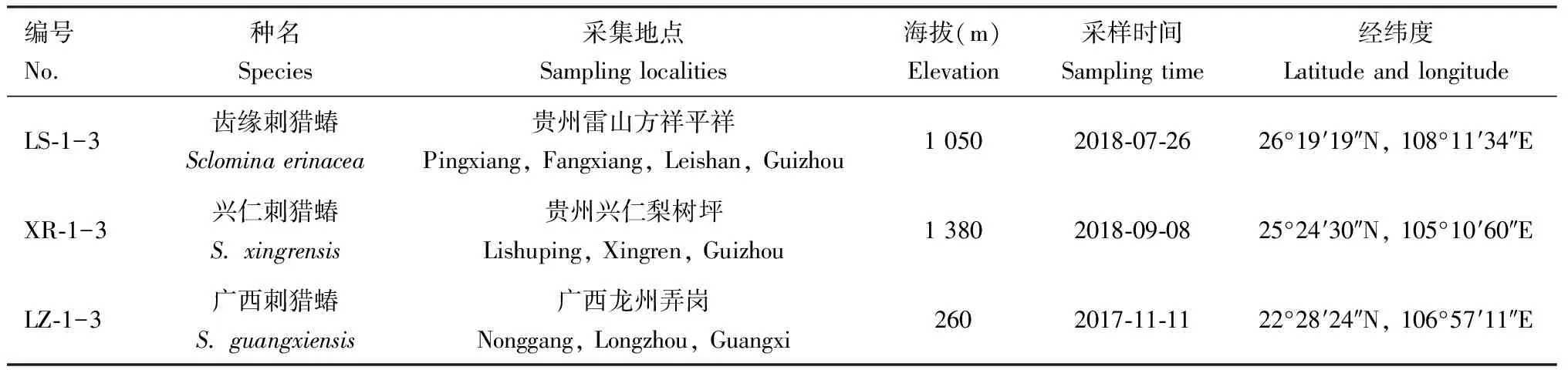
表1 实验材料信息Table 1 Information of experimental materials
1.2 cDNA文库建立及高通量测序
本研究刺猎蝽属3种9样本的转录组测序和数据分析委托广州基迪奥生物科技有限公司完成。利用QiaQuick PCR试剂盒构建cDNA文库,使用Agilent 2100 Bioanalyzer和ABI StepOnePlus Real-Time PCR System质检合格后,通过Illumina HiSeqTM4000进行测序。原始序列下机后,去除含有adaptor、N比例大于10%和低质量的读段,获得高质量序列,使用软件Trinity v2.6.5(Grabherretal., 2011)进行拼接和组装,获得unigene,NCBI序列号:SUB12958005(Submission ID),PRJNA994979 (BioProject ID)。
1.3 生物信息学分析
通过blastx(Altschuletal., 1990)将齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽的转录组unigene比对到4个蛋白数据库Nr, Swiss-Prot, KEGG和COG/KOG(E-value<10-5),得到具有最高序列相似性的蛋白,从而得到该unigene的蛋白功能注释信息、Pathway注释、COG/KOG功能注释、GO功能注释等。
1.4 差异表达基因的筛选条件和方法
本研究使用edge R (Robinsonetal., 2010)进行齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽种间差异表达基因分析统计。
使用每千个碱基的转录每百万映射读取的片段(reads/fragments per kilobase per million reads/fragments, RPKM/FPKM)计算unigene的表达量(Mortazavietal., 2008),其计算公式为:RPKM/FPKM=(1 000 000×C)/(N×L/1 000),C为比对到基因的读段数,N为比对到所有基因的总读段数,L为基因的碱基数。
采用FPKM作为衡量差异表达基因表达量的指标(Mortazavietal., 2008),利用错误发现率(false discovery rate, FDR)与log2FC来筛选差异表达基因,筛选条件为FDR<0.05 且|log2FC|>1(即:差异表达基因表达量的变化倍数>2)(Benjamini and Hochberg, 1995)。前者差异表达基因的表达量高于后者,即两者基因表达量的比值,以2为底求对数值,如果结果是正值(即差异表达基因表达量的变化倍数需要2倍以上),则种间差异表达基因为上调基因(up-regulated gene),否则为下调基因(down-regulated gene),累计上调或下调的差异表达基因数,表现在差异表达基因统计图中,红色表达量上调和绿色表达量下调,没有差异不统计。得到差异表达基因之后,进行GO功能注释和KEGG通路富集分析。
1.5 差异表达基因表达模式聚类
本研究基于基因表达量,对样本和基因间的关系进行层级聚类,并使用热图来呈现聚类结果。首先对齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽9个样本的转录组基因表达量以2为底求对数值,然后对不同样品和基因进行层级聚类分析,热图中每行代表1个样品,每列代表1个基因,基因在不同样品中的表达量用不同颜色表示。红色为表达量上调,颜色越红表示表达量越高;蓝色为表达量下调,颜色越蓝表示表达量越低。
另外,进攻龙游的鬼子今天刚得手,骄兵必松。加上这一带又是你们二十八军的游击区,假如遭遇上鬼子没准还多一些照应。”
2 结果
2.1 转录组测序结果统计
将齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽转录组测序原始数据经过过滤后,Q20碱基百分比在98%以上,Q30碱基百分比在95%以上,GC含量在33.80%~36.01%之间,这表明所测得数据准确,可以用于后期组装,也满足后续分析的要求(表2)。

表2 齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽转录组测序数据统计Table 2 Transcriptome sequencing data statistics of Sclomina erinacea, S. xingrensis and S. guangxiensis
组装获得42 215个unigenes,长度为28 599 724 bp,N50长度为1 024 bp,G+C含量约为36.72%,其中,最大unigene的长度为14 702 bp,最小unigene长度为201 bp,平均长度为677 bp。N50unigene长度为1 024 bp,拼接组装结果良好。
通过对齿缘刺猎蝽、广西刺猎蝽和兴仁刺猎蝽3种刺猎蝽属昆虫的9个样本进行转录组测序,基于全体基因表达量,对样本间的关系进行层级聚类,得到样本间的层级聚类关系(图1),将不同样本中表达模式相同或相似的基因聚为一类,表达模式相似的基因可能具有相似的功能,共同参与同一代谢过程或存在于同一细胞通路中。刺猎蝽属的3个种在热图上反映了样本间的转录组差异表达,聚类图上各地样本基本一致(仅齿缘刺猎蝽的雷山样本LS-2重复性出现不一致),组内样本间的表达模式相似,说明组内样本间差异较小。图1中的XR-1-3与LZ-1-3两个组之间存在着很大的差异,XR-1-3左侧组基因大多呈现上调,而右侧组呈现下调;LZ-1-3与XR-1-3相反。LS-1和LS-3与其他两种刺猎蝽样本有关系,在同位置均出现上调和下调,但不如其他两种之间明显。
2.2 转录组基因功能注释
齿缘刺猎蝽、广西刺猎蝽和兴仁刺猎蝽的转录组共有21 117个基因在4个蛋白质数据库中得到注释,占比50.02%(图2),其中,Nr数据库注释到的基因数最多,为20 522个(图2, 3),其次是Swiss-Prot数据库注释到15 550个基因(图2),再次是COG/KOG数据库注释到13 969个基因(图2, 4),注释基因数最少的是KEGG数据库(图2, 5),为10 850个。4个数据库共同注释到的基因共有9 556个(图2)。仅在Nr数据库中被注释到的基因数最多,为4 385个;其次依次为Swiss-Prot,425个,COG/KOG,27个,KEGG, 11个(图2)。

图2 齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽转录组基因功能数据库注释韦恩图Fig. 2 Venn diagrams of database function annotations for genes in the transcriptomes of Sclomina erinacea,S. xingrensis and S. guangxiensis
Nr数据库20 522个unigenes的GO功能注释结果表明(图3):刺猎蝽属转录组基因功能分为生物学过程(biological process)、细胞组分(cellular component)和分子功能(molecular function)三大类、56个功能亚类,注释到生物学过程有23个功能亚类,注释到细胞成分的21个功能亚类,注释到分子功能的12个功能亚类。在生物学过程大类中注释到代谢过程(metabolic process)功能亚类的基因最多;在细胞组分大类中注释到细胞(cell)和细胞部分 (cell part)功能亚类的基因最多;在分子功能大类中注释到结合(binding)功能亚类和催化活性(catalytic activity)的基因数最多。
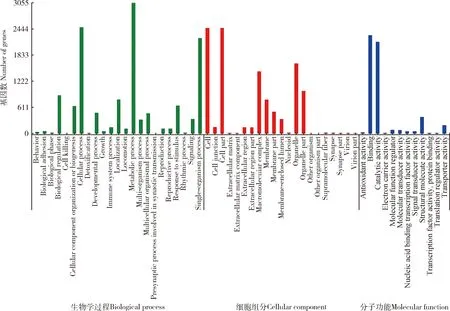
图3 齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽转录组基因的GO功能注释Fig. 3 GO functional annotations of genes in the transcriptomes of Sclomina erinacea, S. xingrensis and S. guangxiensis
齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽转录组的42 215个unigenes均经过COG/KOG数据库的功能预测和分类,共有13 969个基因得到注释(图4),并被分为25个COG/KOG类(缺少X类),其中,共有4个COG/KOG类(J, O, R, T)包含2 000条以上的unigenes(图4),仅一般功能预测(general function prediction only, R),超过4 000条基因,占比最多;其次是信号转导机制(signal transduction mechanisms, T),超过3 000条基因;再次是翻译后修饰、蛋白质周转和分子伴侣(posttranslational modification, protein turnover, chaperones, O);最后是翻译、核糖体结构和生物发生(translation, ribosomal structure and biogenesis, J),其余COG/KOG注释情况如图4所示。
齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽转录组共有10 850个unigenes成功注释到KEGG数据库,涉及到32个KEGG通路(图5),其中,被注释到的unigenes基因数超过1 000的通路包括:参与新陈代谢系统中全局和总览(global and overview)通路的unigenes最多,为4 873个;其次依次是翻译(translation)通路,1 866个,碳水化合物代谢(carbohydrate metabolism)通路,1 477个,及氨基酸代谢(amino acid metabolism)通路,1 212个。除此之外,824个unigenes参与到了运输和分解代谢(transport and catabolism)通路中,其余通路unigenes基因数如图5所示。
2.3 种间差异表达基因
由图6可知,兴仁刺猎蝽vs齿缘刺猎蝽转录组中表达量上调的基因有803个,表达量下调的基因有2 587个,差异基因表达总数3 390个;兴仁刺猎蝽vs广西刺猎蝽转录组中表达量上调的基因有6 639个,表达量下调的基因有5 904个,差异表达基因总数12 543个;齿缘刺猎蝽vs广西刺猎蝽转录组中表达量上调的基因有2 485个,表达量下调的基因有1 095个,差异表达基因总数3 580个。3组间比较,兴仁刺猎蝽vs广西刺猎蝽转录组中的上调和下调基因数量总和最大,其他两组上下调基因数量总和接近。
2.4 种间差异表达基因GO功能注释
将兴仁刺猎蝽vs齿缘刺猎蝽、兴仁刺猎蝽vs广西刺猎蝽、齿缘刺猎蝽vs广西刺猎蝽的转录组差异表达基因进行GO功能注释,结果如图7-9所示,差异表达基因被分类到分子功能、细胞组分和生物学过程三大类及各个功能亚类中。
由图7可知,兴仁刺猎蝽vs齿缘刺猎蝽转录组中差异表达基因从高到低分别被注释到生物学过程、细胞组分和分子功能的代谢过程、细胞、细胞部分等50个功能亚类中;在生物学过程这一大类中,差异表达基因中注释到代谢过程、细胞过程(cellular process)和单生物过程(single-organism process)在20个功能亚类中基因数较高;在细胞组分功能这一大类中,差异表达基因中注释到细胞、细胞部分、大分子复合体(macromolecular complex)和细胞器(organelle)在19个功能亚类中所占基因数较高;在分子功能这一大类中,差异表达基因中注释到结合(binding)和催化活性(catalytic activity)在11个功能亚类基因数较高。该组对比中,兴仁刺猎蝽vs齿缘刺猎蝽的转录组中差异表达基因明显下调,少数基因上调。
由图8可知,兴仁刺猎蝽vs广西刺猎蝽转录组中的差异表达基因数从高到低依次分别被注释到生物学过程、细胞部分和细胞等54个功能亚类中;在生物学过程这一大类中,差异表达基因中注释到代谢过程、细胞过程和单生物过程在21个功能亚类中基因数较高;在细胞组分功能这一大类中,差异表达基因中注释到细胞、细胞部分、大分子复合体和细胞器在21个功能亚类中基因数较高;在分子功能这一大类中,差异表达基因中注释到结合和催化活性在12个功能亚类中基因数较高。该组对比中,兴仁刺猎蝽vs广西刺猎蝽转录组中差异表达基因存在明显上调和下调。
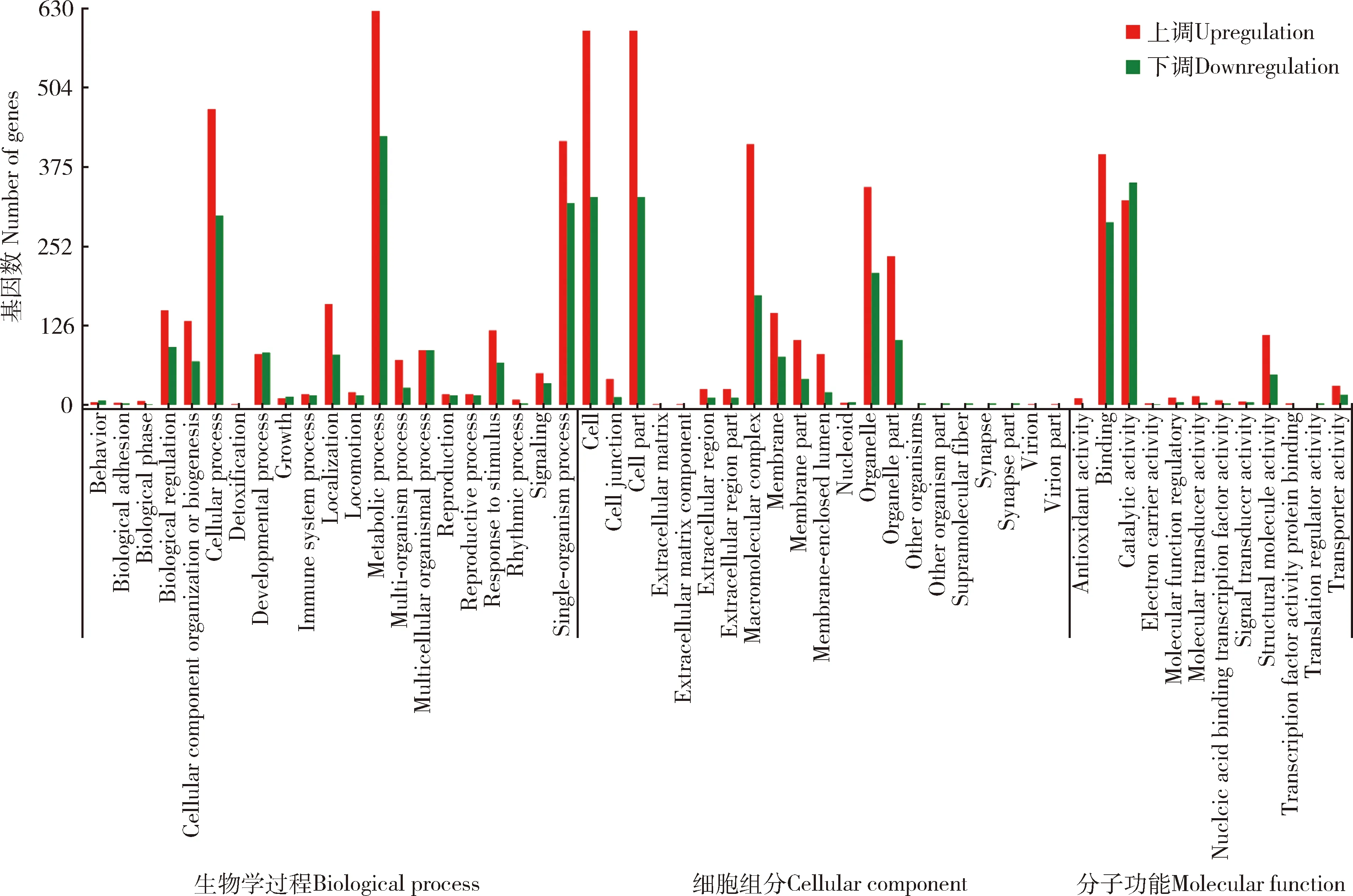
图8 兴仁刺猎蝽vs广西刺猎蝽转录组中差异表达基因的GO功能注释Fig. 8 GO functional annotation of differentially expressed genes in the Sclomina xingrensis vs S. guangxiensis transcriptomes
由图9可知,齿缘刺猎蝽vs广西刺猎蝽转录组差异表达基因被分别注释到生物学过程、细胞组分和分子功能中细胞、细胞部分和代谢过程等46个功能亚类中。在生物学过程这一大类中,差异表达基因中注释到代谢过程和细胞过程在20个功能亚类中所占比例较高;在细胞组分功能这一大类中,差异表达基因中注释到细胞和细胞部分在14个功能亚类中所占比例较高;在分子功能这一大类中,差异表达基因中注释到结合和催化活性2个功能亚类中占比较高。该组对比中,转录组中差异表达基因明显上调,略下调。
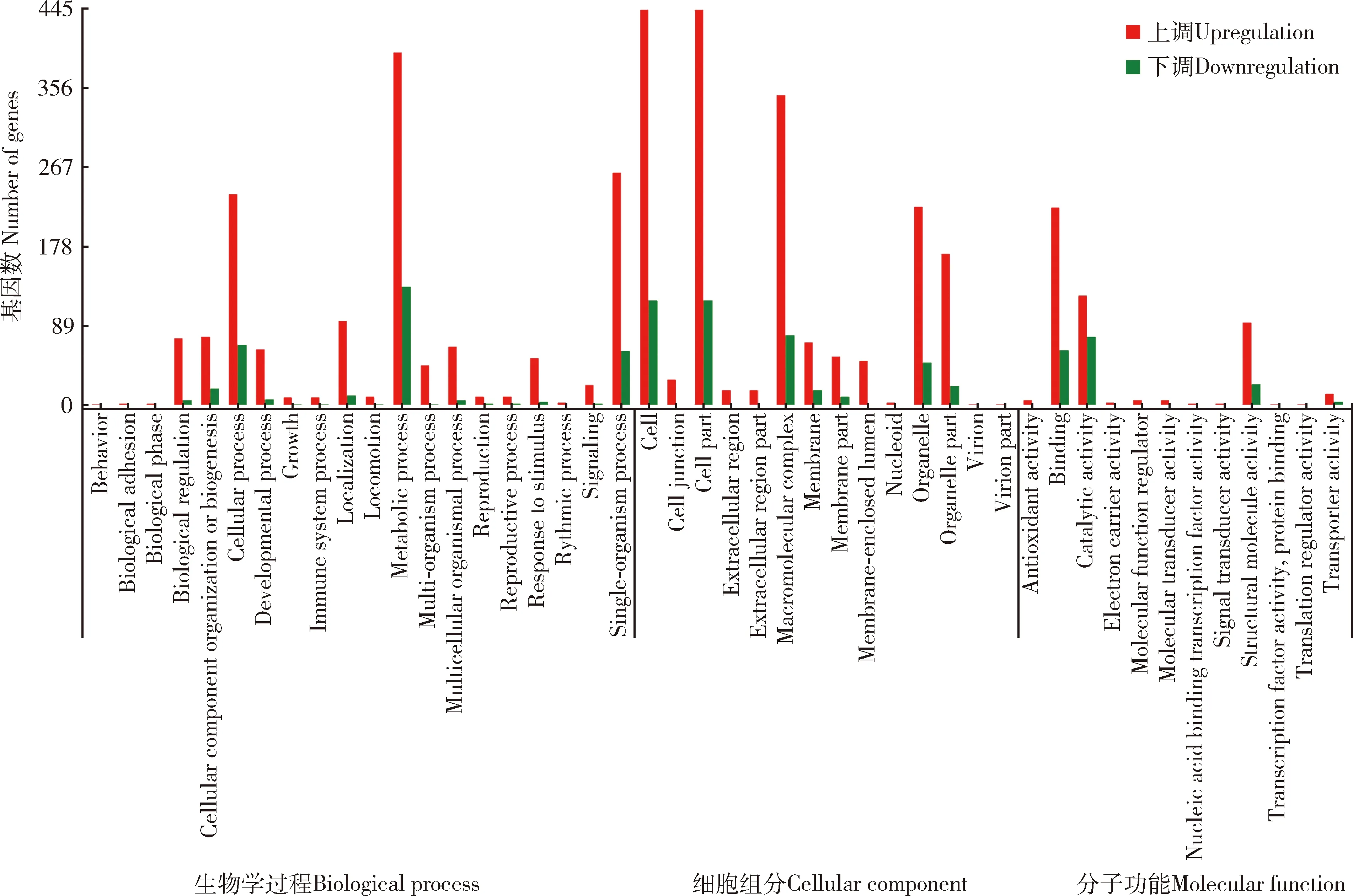
图9 齿缘刺猎蝽vs广西刺猎蝽转录组中差异表达基因的GO功能注释Fig. 9 GO functional annotation of differentially expressed genes in the Sclomina erinacea vs S. guangxiensis transcriptomes
2.5 种间差异表达基因KEGG通路富集
齿缘刺猎蝽、兴仁刺猎蝽和广西刺猎蝽转录组的6 254个差异表达基因参与多个不同的KEGG代谢通路,包括代谢通路(metabolic pathways)、药物代谢-细胞色素P450(drug metabolism-cytochrome P450)、不同环境下微生物代谢(microbial metabolism in diverse environments)、糖酵解/糖异生(glycolysis/gluconeogenesis)、脂肪酸降解(fatty acid degradation)、过氧化物酶体(peroxisome)、 芳香族化合物降解(degradation of aromatic compounds)和酪氨酸代谢(tyrosine metabolism)等(表3-5)。

表3 兴仁刺猎蝽vs齿缘刺猎蝽转录组中差异表达基因(DEGs) KEGG通路富集(前10)Table 3 KEGG pathway enrichment of differentially expressed genes (DEGs)in the Sclomina xingrensis vs S. erinacea transcriptomes (Top 10)
兴仁刺猎蝽vs齿缘刺猎蝽转录组差异表达基因富集到164个KEGG代谢通路,被注释的差异表达基因702个(表3)。兴仁刺猎蝽vs广西刺猎蝽转录组差异表达基因富集到201个KEGG通路,被注释的差异表达基因2 091个(表4)。齿缘刺猎蝽vs广西刺猎蝽转录组差异表达基因富集到129个KEGG代谢通路,被注释的差异表达基因865个(表5)。

表4 兴仁刺猎蝽vs广西刺猎蝽转录组中差异表达基因(DEGs) KEGG 通路富集(前10)Table 4 KEGG pathway enrichment of differentially expressed genes (DEGs) inthe Sclomina xingrensis vs S. guangxiensis transcriptomes (Top 10)
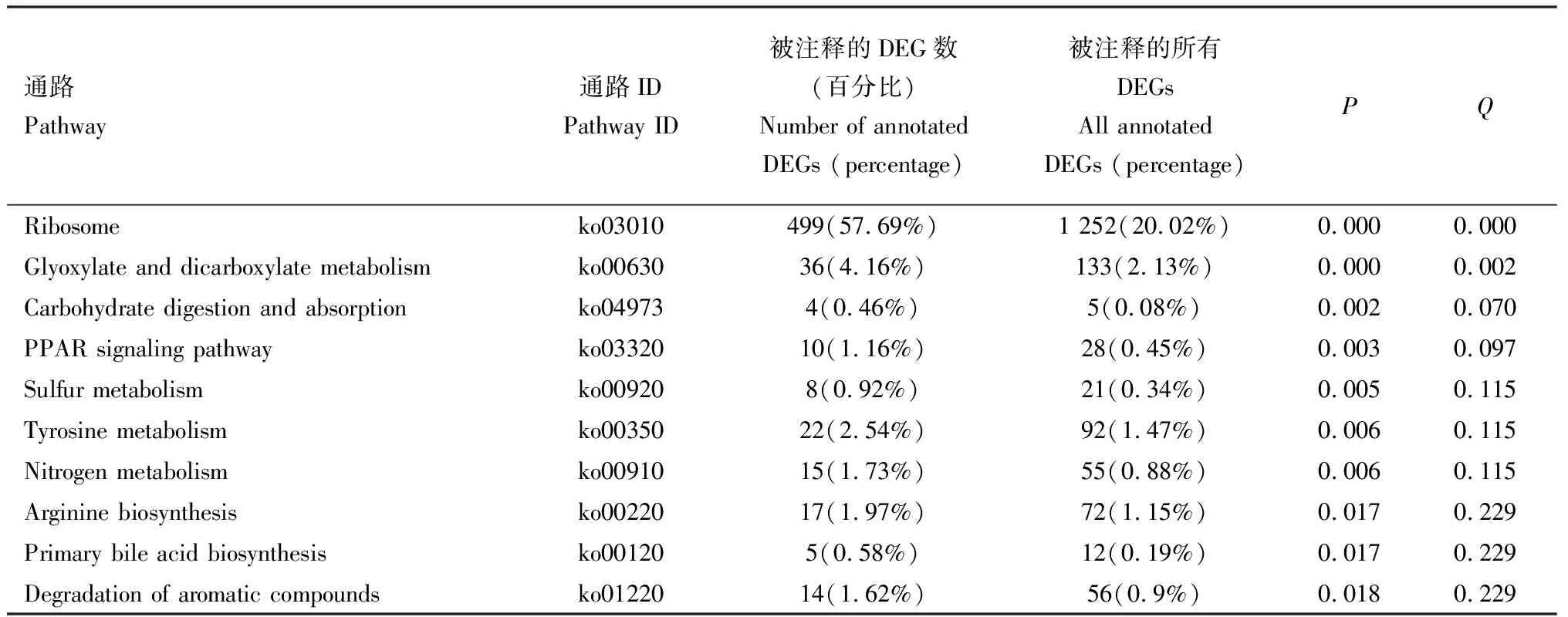
表5 齿缘刺猎蝽vs 广西刺猎蝽转录组中差异表达基因(DEGs) KEGG 通路富集(前10)Table 5 KEGG pathway enrichment of differentially expressed genes (DEGs) inthe Sclomina erinacea vs S. guangxiensis transcriptomes (Top 10)
其中,有1 995个差异表达基因富集到代谢通路(metabolic pathways),占富集基因总数的31.9%。兴仁刺猎蝽vs齿缘刺猎蝽转录组中有349个差异表达基因富集到代谢通路,兴仁刺猎蝽vs广西刺猎蝽转录组中有764个,齿缘刺猎蝽vs广西刺猎蝽转录组中有221个。
有109个差异表达基因富集到药物代谢-细胞色素P450,占富集基因总数的1.74%。兴仁刺猎蝽vs齿缘刺猎蝽转录组中有26个差异表达基因富集到药物代谢-细胞色素P450通路,兴仁刺猎蝽vs广西刺猎蝽转录组中有57个,齿缘刺猎蝽vs广西刺猎蝽转录组中有15个。
有1 252个差异表达基因富集到核糖体(ribosome)通路,占富集基因总数的20.02%。兴仁刺猎蝽vs齿缘刺猎蝽转录组中有171个差异表达基因富集到核糖体通路,兴仁刺猎蝽vs广西刺猎蝽转录组中有610个,齿缘刺猎蝽vs广西刺猎蝽转录组中有499个。
3 讨论
转录组测序以N50、Q30来评估转录组数据的质量。一般认为N50值越大就表明得到的长片段越多(贾新平等, 2014),且Q30值大于85%说明测序结果可靠(Jinetal., 2016)。通过对齿缘刺猎蝽、广西刺猎蝽和兴仁刺猎蝽进行转录组测序(表2),共获得unigene序列42 215个,平均长677 bp,N50长1 024 bp,Q30值都大于85%。从N50长度和Q30值等方面均显示本次拼接结果良好。将拼接好的unigene序列放入四大数据库进行功能注释,结果表明,21 117个unigenes (50.02%) 被成功注释,剩余的21 098个unigenes(49.98%) 未被成功注释。未被注释的原因可能是unigene 较短,未与公共数据库中的序列比对上(孟翔等, 2016),也有可能是存在很多功能未知的新基因(Houetal., 2011)。
通过种间的层级聚类关系(图1)分析兴仁刺猎蝽、齿缘刺猎蝽和广西刺猎蝽的每个基因的表达量情况,同种表现出相近的基因表达模式;不同种的表达模式相同或相似的基因聚为一类,表达模式相似的基因可能具有相似的功能,共同参与同一代谢过程或存在于同一细胞通路中,为今后进一步研究奠定基础。
生物信息学分析表明 (图2-5),Nr 数据库的GO注释到生物学过程基因数量最多,注释到细胞组分的次之,注释到分子功能的最少。COG/KOG注释结果表明(图4): 42 215个基因中有13 969个得到注释,被分为 25个COG/KOG功能类,其中,共有4个COG/KOG功能类包含2 000个以上的基因,分别是:仅一般功能预测、信号转导机制、翻译后修饰、蛋白质周转和分子伴侣和翻译、核糖体结构和生物发生。KEGG注释结果表明(图5):共有10 850个基因注释到KEGG数据库,其中参与新陈代谢系统中全局和总览的最多。
种间差异表达基因分析发现(图6),兴仁刺猎蝽vs齿缘刺猎蝽的差异表达基因3 390个,上调803个,下调2 587个,主要表现在下调;兴仁刺猎蝽vs广西刺猎蝽的差异表达基因12 543个,上调基因数为6 639,下调基因数5 904,上调和下调均较多;齿缘刺猎蝽vs广西刺猎蝽的差异表达基因3 580个,上调基因数2 485,下调的基因数1 095,主要表现在上调。3组间比较,兴仁刺猎蝽vs广西刺猎蝽的上调和下调基因数量总和最大,体现出兴仁刺猎蝽与广西刺猎蝽的亲缘关系较远,与基于DNA 条形码COI序列数据的系统发育和物种界定结果(Zhaoetal., 2021)一致。其他两组上下调基因数量总和接近。通过上述差异比较,由基因层面也进一步支持了齿缘刺猎蝽、广西刺猎蝽和兴仁刺猎蝽的遗传差异。
兴仁刺猎蝽vs齿缘刺猎蝽,兴仁刺猎蝽vs广西刺猎蝽和齿缘刺猎蝽vs广西刺猎蝽转录组中差异表达基因KEGG通路显著富集分析表明(表3-5):通过KEGG代谢通路分析共得到6 254个差异表达基因,各种的差异表达基因参与不同的代谢通路。兴仁刺猎蝽vs广西刺猎蝽转录组中富集到KEGG通路差异表达基因数最高,兴仁刺猎蝽vs齿缘刺猎蝽和齿缘刺猎蝽vs广西刺猎蝽转录组中富集到KEGG通路差异表达基因数不确定。
刺猎属昆虫是广泛分布在我国南方地区特有的森林生态系统中常见的天敌昆虫,它们随着地理分布改变,各地居群分化显著,是研究中国昆虫物种分化的理想材料,刺猎蝽属与悬钩子属植物保持着紧密的关系,但是还有更多未知进化生存策略不为人们所知。本研究利用高通量测序技术获得刺猎蝽属的转录组数据,为后续探索该属物种的进化规律奠定了分子基础。
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
草业学报(2022年3期)2022-03-26 02:27:28
传染病信息(2021年6期)2021-02-12 01:52:58
晚晴(2021年12期)2021-01-10 19:03:04
四川地质学报(2020年3期)2020-05-22 06:25:48
中国实验诊断学(2017年5期)2017-06-05 15:03:23
艺术评鉴(2016年3期)2016-05-30 10:48:04
传奇·传记文学选刊(2015年1期)2015-05-30 06:00:02
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
化学工业与工程(2015年1期)2015-02-10 03:01:41
