
复方制剂有关物质研究及其对产品优化的作用
2023-10-27 08:13:02唐丽莉黄慧萍
当代化工研究 2023年19期
*唐丽莉 黄慧萍
(复旦大学 上海 200433)
制药从业者在研究和生产药品时,既希望药品有效,又希望能减少其毒副作用。药品的毒副作用不仅来源于主药的药理活性,还来源于药品中存在的杂质。因此,杂质控制是药品安全性研究的核心内容。药品中的杂质通常分为:有机杂质、无机杂质、残留溶剂。有机杂质可在药品的生产或贮存期间产生,也可由药物与辅料或包装结构的相互作用产生,其中化学结构与活性成分类似或具渊源关系的有机杂质,通常称为有关物质。根据中国药典药品杂质分析指导原则的规定,除降解产物和毒性杂质外,原料药中已控制的杂质,制剂中一般不再控制,制剂杂质研究重点在于降解产物。
复方制剂系指含有多个主药成分(≥2个)的制剂,由于其主药成分较多,复方制剂相较于单方制剂的杂质研究更为复杂,不仅包括每个原料药中的杂质,还包括生产和贮存期间产生的降解产物以及主药与主药、主药与辅料之间相互作用产生的降解产物。对于各原料带入的杂质,原料药供应商均已进行了全面的研究,制剂研究可加以参考。复方制剂杂质研究的难点在于降解产物。对于复方制剂降解产物的研究,国内外很少涉及,而基于复方制剂有关物质研究优化其产品和改进工艺的案例则更少之甚少。
本文将重点阐述复方制剂有关物质研究的基本思路,并将其与产品优化的实际案例相结合,介绍对复方制剂有关物质研究和产品优化的整体方法与基本思路,供复方制剂有关物质研究和产品优化工作参考。
1.杂质分析预测
单方制剂和复方制剂杂质研究过程中需要重点关注的杂质均为降解产物。复方制剂相对单方制剂而言,由于其原料数量为两个及以上,原料之间也会由于相互作用而产生降解产物。因此,复方制剂不仅要从原料药和单方制剂的稳定性实验结果来预测杂质,还需基于各原料药的化学结构等方面开展分析,以此预测复方制剂中可能存在的降解产物。此类预测的方法有很多,较为传统的方法是通过药物的结构来分析两个药物之间是否会存在相互作用及可能的反应类型。此外,还可依托模型来分析两个药物之间的相互作用,如:PBPK模型等。
2.建立复方制剂有关物质检测方法
复方制剂的杂质数量和种类纷繁复杂。如何开发一个有效的分析方法对有关物质进行检测,是复方制剂有关物质研究中最难且最为关键的一步。笔者建议通过如下方式建立复方制剂有关物质的检测方法。
(1)初步检测条件建立。查询各国药典(如:美国药典USP、欧洲药典EP、日本药典JP、中国药典Ch.P等),汇总并比较各国药典中收录的各主成分杂质检测方法,同时收集各国药典中收载的各主成分杂质作为后续杂质结构确证及定位的基础。
基于前期预测的杂质,综合评估各杂质的分离情况,从各国药典主成分有关物质检测方法中,选取一个相对较为合适的方法作为有关物质检测的初始条件,考虑因素包含:方法可以检测出较多杂质或者方法的合理性等。由于复方制剂涉及杂质较多且分离较难,正相液相色谱法较难实现有效分离,通常会选用反相液相色谱技术,通过梯度洗脱来检测复方制剂的有关物质。
(2)分析方法优化。从初始色谱检测条件出发开展优化,直至找出可以有效分离各个已知杂质和未知杂质的色谱条件。一般情况下,杂质初步分离的色谱条件还不足以开展杂质的定量检测,可能存在主峰与杂质峰分离度不理想的情况,或者,在一段时间内无杂质检出等问题。后续,应分别从洗脱梯度、色谱柱筛选、流动相体系、缓冲盐、柱温、检测波长等方面摸索出最为适宜的检测条件。这就需要分析人员具有充足的经验,摸索出最为适宜的分析方法。
(3)分析方法验证。对新开发的检测方法进行分析方法验证,从专属性、检测限、定量限、线性、准确性、范围、精密度、耐用性等方面,确证新开发的检测方法适用于复方制剂产品的检测。
3.杂质谱研究
在明确初步色谱条件后,通过对复方制剂近效期产品的检测,获得代表批次色谱图。结合原料药供应商的杂质研究结果和各国药典中收载的各主成分杂质,对色谱图进行分析,将已知杂质定位,并标记未知杂质。在分析未知杂质时,可以采用HPLC-MS联用法,通过HPLC-MS对HPLC图谱中出现的未知色谱峰进行质谱分析,得到未知杂质的质谱图,根据未知杂质的分子离子峰和其裂解后产生的碎片离子峰的信息进行初步的质谱分析,从而推测该杂质与哪种主成分有关。需特别需注意的是,由于色谱处理也可能对复方制剂杂质产生影响,某些色谱峰中出现的杂质,可能在实际生产条件下未必生成,比如:某个杂质为原料水解开环后的产物,该杂质只能在强碱条件下才能保持相对稳定,在弱碱条件下,会闭环回到原先结构,所以,对于这些类型的杂质,较难通过制备分离获得,而是否需进一步对其进行管控,就要结合具体处方和工艺开展评估,确定在现有处方和工艺条件下,是否有可能产生此类杂质,根据评估结果,明确具体的管控措施,比如:某杂质在强碱条件下稳定在弱碱条件下回到原先结构,而实际生产过程未使用到强碱类的物料,因此,可在适当评估后略去对该杂质的研究。
在此期间,还应关注复方制剂产品各组分含量变化平均值和有关物质平均总检出量,明确有关物质生成量和其含量下降量之间是否能符合质量守恒,从而再次确证复方制剂有关物质检测方法的适用性。
4.杂质归属研究
根据ICH新制剂中的杂质(Q3B)与《中国药典)(2020年版)9102药品杂质分析指导原则,对于表观含量在鉴定阈值及以上的单个杂质和在鉴定阈值以下但具强烈生物作用的单个杂质或毒性杂质,予以定性或确证其结构。

表1 制剂的杂质限度
杂质归属的研究手段较多,主要包含HPLC图谱比对,光、热、湿影响因素试验以及稳定性加速试验的HPLC图谱比较分析。本文将重点阐述如何通过光、热、湿影响因素试验来研究复方制剂的降解产物,并以含4种主成分的复方制剂为例,阐述配伍设计的基本方法。
假设某复方制剂共有4种原料药和若干辅料,其配伍设计参考如下。
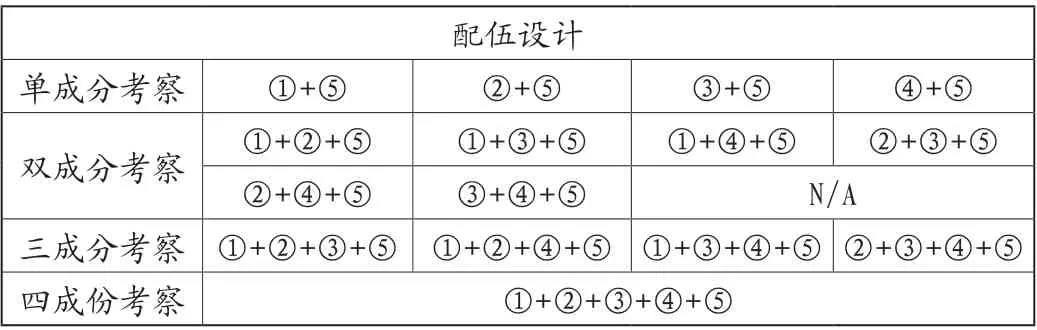
表2 配伍设计
研究时,应逐一列出各个主药在单成份、双成份、三成份和四成份及其于不同强制降解条件下的杂质及其含量。在完成所有的影响因素试验后。重点关注于在鉴定限度以上的杂质,汇总分析其在何种条件下,以哪种配伍形式生成。并结合原料药和杂质的物理和化学特性,分析该杂质生成的机理。
在进行杂质归属分析时,还应汇总所关注杂质在不同效期的杂质含量及其生成速率,有些杂质的生成会呈现一定的相关性,一般可推测为两个主成分相互作用下产生的两个杂质,如:含有一对孤对电子的主成分进攻具有亲电性缺电子中心的另一个主成分,从而发生去甲基/甲基化等相互作用,形成两个呈现一定相关性的杂质。主药之间的相互作用方式有很多,本文列举一二仅做示例。
此外,杂质归属要结合实际生产工艺和贮存条件综合考虑,比如,在杂质归属实验中发现某个杂质在高温条件下由两个主药配伍时生成。如遇到这种情况,在分析时要评估内容包括:①高温条件对其影响更为显著,还是两个主药配伍对其影响更为显著;②在实际生产中,这两个主药配伍紧密性如何;③产品的储运条件是否有所规定,以上几个因素都对杂质归属研究存在一定的指导意义和确证作用,并对后续杂质改进提供很大的参考价值。
5.产品优化
(1)基本思路。基于杂质生成的机理,可以通过诸多方式优化产品,如:变更产品贮藏条件、优化产品生产工艺、变更原辅料来源、改变产品的包装材料或包装形式等。
如在研究中发现,某个有关物质的生成机理为主药与主药之间的相互作用,或者主药与辅料之间的相互作用,可考虑通过工艺改进来降低有关物质,例如,调节主药投料顺序等。由于该方式涉及工艺变更,需要按照《已上市化学药品药学变更研究技术指导原则》开展相应的研究和验证工作,并按规定申报或备案。
其他还有许多改善产品的方式,例如,改变湿法制粒为干法制粒,改变原料药和辅料的来源和种类、改变包装材料等。本文不再赘述。总之,应基于前期研究,采取合适的改善措施,并加以确证。
(2)案例分析。本文将根据笔者实际经历的研究,侧重于阐述如何基于杂质生成机理,从改变产品贮藏条件和优化产品工艺这两种方式来提高产品质量。
某复方制剂中两个原料药所属有关物质的生成呈相关性,原料药1的有关物质随着原料药2的有关物质的生成不断增加。在该复方制剂的强制降解试验中,显示其是在原料药1和原料药2混合辅料配伍时在高温条件下生成。因此,从前期的杂质谱分析和强制降解试验,并结合两个原料药及其有关物质结构分析,推测出这两个有关物质的生成机理为:①原料药1的甲氧基在有氢离子存在的条件下形成缺电子中心原子,具有亲电性;原料药2的N,N-二甲基的N含有一对孤对电子(亲核试剂)进攻具有亲电性的甲基缺电子中心,从而生成原料药2的N,N-二甲基N位的甲基化杂质(原料药2有关物质)和去甲基化杂质(原料药1有关物质)。
基于杂质生成机理,考虑以两种方式开展优化。
①变更产品的贮藏条件:原产品的贮藏条件为常温,根据强制降解试验显示,该产品对温度较为敏感。将贮藏条件变更为阴凉,并基于变更后的贮藏条件,对复方制剂开展长期留样观察至产品有效期,获得相关数据并进行汇总分析后发现,两个原料药相互作用所产生的有关物质均得到了有效的控制。
②优化产品生产工艺:经调查发现,这两个存在相互作用的原料药均在同一工段同时投料,考虑将该两个原料药分别在不同工段投料,尽可能改善两个原料药配伍过于紧密的问题,并基于优化后工艺,开展稳定性加速试验,统计两个原料药有关物质的情况,发现其均得到了有效的控制。
鉴于两种方法均可对有关物质进行有效控制,结合成本收益考量,选择相对更为合适的方法,经批准后实施。
6.小结与展望
药品不同于普通商品,不仅要有效,还要安全。无论是新药研发还是传统药的优化,杂质研究都是药品质量控制的重中之重。而很多传统药,受限于早期较为落后的技术手段,对杂质的控制并不完善。制药行业从业人员,应以患者的健康为要务,致力于探索更为有效的杂质检测方法,并依托先进有效的检测方法,不断改善药品质量,以一种获益和风险平衡的方式,尽可能降低药品中的杂质,以期提高药品的安全性。
猜你喜欢
云南化工(2021年7期)2021-12-21 07:27:48
艺术品鉴(2020年6期)2020-12-06 10:49:08
中西医结合心血管病电子杂志(2020年5期)2020-06-08 15:49:48
中国盐业(2018年20期)2019-01-14 01:18:42
领导文萃(2017年6期)2017-03-24 09:31:39
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
现代临床医学(2016年2期)2016-02-19 17:51:51
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
中华皮肤科杂志(2014年4期)2014-12-19 12:55:59
