
铜代谢失调与细胞损伤及肝病的关系
2023-10-25 02:20柳涛刘雅丽张飞宇高沿航
临床肝胆病杂志 2023年9期
柳涛, 刘雅丽, 张飞宇, 高沿航
吉林大学第一医院肝胆胰内科, 长春 130021
铜是人体必需的微量元素,为人体许多代谢关键酶的重要组成成分,如:Cu/Zn超氧化物歧化酶1(copper/zinc superoxide dismutase,SOD1)、细胞色素C氧化酶(cytochrome C oxidase,CCO)、赖氨酰氧化酶(lysyl oxidase,LOX),人体在细胞和分子水平上的铜代谢过程见图1。因具有两种离子状态,所以其在机体氧化还原反应中是重要的辅助因子。2022年3月研究人员新发现一种铜依赖性细胞死亡方式:铜死亡(cuprotosis)[1],该发现进一步强调人体对铜稳态机制的需求。人体铜水平处于动态平衡,无论遗传性还是获得性铜稳态失调均会导致或加重某些疾病,如Menkes病、Wilson病(Wilson’s disease,WD)、癌症等。近年来随着研究的不断深入,研究人员发现铜稳态失衡会通过多种途径促进细胞损伤并与多种肝脏疾病的发生、发展密切相关[2-3]。本文总结近年来有关研究结果,为未来肝脏疾病诊疗提供新思路。
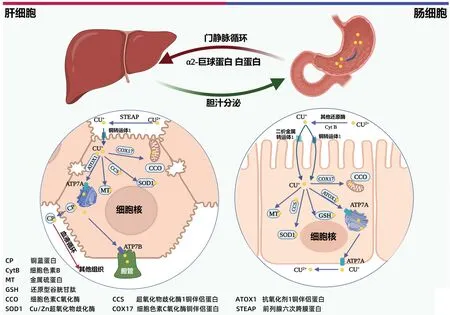
图1 肝细胞的铜稳态机制Figure 1 Mechanism of copper homeostasis in hepatocytes
1 铜稳态失衡促进细胞损伤机制
1.1 铜和氧化应激
活性氧(reactive oxygen species,ROS)是人体细胞代谢副产物,高浓度的ROS可引起细胞结构与功能损伤、增加细胞癌变的风险、诱导细胞死亡的发生[4]。生理状态下胞内铜处于一种动态平衡,铜水平过高或过低均可使细胞发生氧化应激。
1.1.1 细胞内铜超负荷导致氧化应激发生 铜超负荷时可通过Fenton反应产生大量ROS,降低胞内还原型谷胱甘肽(glutathione,GSH)含量,导致细胞对有害刺激敏感性升高。有研究[5]表明胞内铜超负荷通过诱发氧化应激,导致线粒体膜通透性改变、DNA损伤、促凋亡蛋白表达增加[如B细胞淋巴瘤-2基因的相关X蛋白(B cell lymphoma -2 associated X protein,BAX)、半胱天冬酶-3、9],进而诱发细胞凋亡的发生。
1.1.2 细胞内铜缺乏导致氧化应激发生 人体存在多种铜依赖性抗氧化酶(如:SOD1、过氧化氢酶、谷胱甘肽过氧化物酶),铜缺乏导致上述铜酶活性降低,胞内ROS蓄积诱导氧化应激的发生。SOD1突变是肌萎缩性侧索硬化症的重要遗传病因,研究证实神经元细胞内铜缺乏通过增加突变的SOD1错误折叠和改变其疏水性导致神经细胞损伤和死亡的发生,而口服铜络合物可明显减少小鼠脊髓内的运动神经元死亡数量[6]。
1.2 铜与蛋白酶体 研究表明肿瘤细胞内蛋白酶体活性明显增加,通过降解细胞周期蛋白依赖性激酶抑制剂(如P21和P27)、肿瘤抑制因子(如P53)、细胞凋亡抑制因子和细胞凋亡调节因子(如BAX)等促进肿瘤生长。2020年Gałczyńska等[7]研究证实蛋白酶体抑制剂通过促进细胞色素C进入细胞质基质,结合并激活凋亡蛋白酶激活因子1和前体半胱天冬酶-9,导致细胞凋亡的发生。而胞内铜超负荷可通过诱导氧化应激介导26S蛋白酶体的分解进而抑制胞内蛋白酶体活性[8]。
1.3 铜死亡
2022年Tsvetkov等[1]在人类细胞中发现一种不同于已知的细胞死亡方式:铜死亡,这是一种由线粒体铜的靶向积累引发的新形式的细胞死亡,下文将从三个方面简述铜死亡发生机制。
1.3.1 铜代谢紊乱 铜离子载体可以打破人体细胞内铜的动态平衡[1],用铜离子载体(如:伊利司莫)处理后胞内铜水平升高,过多的铜与脂酰化蛋白的硫辛基部分结合诱发脂酰化蛋白聚集,导致细胞铜死亡的发生,而铜螯合剂、铁氧还蛋白1(铜离子载体伊利司莫作用靶点)[9]和硫辛基合成酶基因敲除可以降低细胞发生铜死亡的风险。
1.3.2 三羧酸循环脂酰蛋白功能紊乱 脂酰化蛋白聚集是铜死亡发生的关键,已知蛋白脂酰化仅发生在三羧酸循环中的四种关键酶,丙酮酸脱氢酶复合体的E2亚基二氢硫辛酸转乙酰化酶(dihydrolipoamide transacetylas,DLAT)正是其中之一。铜超负荷引起DLAT的聚集、Fe-S簇蛋白水平降低、热休克蛋白70数量增加、蛋白质毒性应激,从而导致铜死亡的发生。
1.3.3 线粒体有氧呼吸调节铜死亡 线粒体有氧呼吸在铜死亡的发生中起重要调节作用[1],实验证实在缺氧条件下(1% O2)生长的细胞不易发生铜死亡。使用低氧诱导因子(hypoxia-inducible factor-1,HIF-1)分解关键酶脯氨酸羟化酶抑制剂处理常氧条件下(21% O2)生长的细胞,其铜死亡发生率未见明显改变。Tsvetkov等[1]也通过抑制细胞线粒体呼吸证实线粒体有氧呼吸在铜死亡中发挥重要调节作用。
1.4 铜与血管形成 人体血管生成是血管内皮生长因子、碱性成纤维细胞生长因子、IL-1、IL-6、IL-8等多种小分子综合作用的结果[10]。有研究[11]表明,铜可通过与血管生成素直接结合或结合HIF-1后激活上述小分子进而刺激血管生成。此外铜还可通过促进血管内皮增殖及迁移、细胞外基质降解以促进血管生成,胞内铜降低可抑制血管生成,切断组织的营养供应,导致细胞发生损伤和死亡。
1.5 铜与细胞凋亡、自噬、铁死亡 X连锁细胞凋亡抑制蛋白(X-linked inhibitor of apoptosis protein,XIAP)可结合并抑制半胱天冬酶3、7和9的活性进而调节细胞凋亡的发生[12]。XIAP具有很强的铜亲和力,与铜可逆性结合后极易发生泛素化蛋白酶体降解同时丧失抑制细胞凋亡能力,进而降低细胞凋亡的发生阈值。XIAP可通过介导铜代谢MURR1结构域1(copper metabolism MURR1 domain1,COMMD1)的泛素化蛋白酶体降解调节细胞铜水平,铜超负荷可降低胞内XIAP水平,导致COMMD1的水平升高,促进铜从细胞流出[13]。
研究发现胞内异常升高的铜可激活自噬相关的丝氨酸/苏氨酸激酶Unc51样自噬激活激酶1和2(unc-51 like autophagy activating kinase 1、2,ULK1、2)依赖性信号转导通路诱导细胞发生自噬。Yang等[14]研究发现胞内铜超负荷可促进胞内自噬相关基因的转录和翻译,从而抑制铜诱导细胞凋亡的发生。有研究证实ATP7B敲除细胞内自噬相关基因表达水平明显上调[15],Polishchuk等[16]同样发现在WD患者及ATP7B缺乏小鼠肝细胞内过量的铜通过Cu-mTORC1-TFEB信号转导提高胞内自噬水平,抑制自噬可明显加速细胞凋亡的发生。
既往研究发现铁死亡在双硫仑-Cu的抗肿瘤作用中发挥关键作用,Gao等[17]用伊利司莫处理细胞使胞内线粒体铜水平升高,诱导氧化应激与铁死亡的发生。有趣的是,Li等[18]研究证实细胞铜耗竭通过降低胞内GSH含量及抑制谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)活性,也可促进细胞铁死亡的发生,从而建立了铜耗竭与铁死亡的直接联系。除上述的直接联系,Guo等[19]发现胞内铜超负荷通过ROS依赖性AMPK-mTOR途径诱导自噬进而抑制细胞凋亡的发生,而自噬水平升高反过来通过抑制GPX4和降解FTH1等铁蛋白促进细胞铁死亡的发生。但也有研究[20-21]表明肝细胞癌(HCC)细胞内铜超负荷,一方面促进铜蓝蛋白(ceruloplasmin,CP)合成降低胞内铁水平,其次通过Cu-HIF1α-SLC7A11(GSH合成关键蛋白)途径促进GSH合成,进而抑制胞内铁死亡的发生。
线粒体损伤在WD发生发展中发挥重要作用,Zischka等[22]发现在WD早期,随着肝铜水平升高引起线粒体渐进式结构损伤,而这早于氧化应激的发生。胞内铜水平升高及线粒体损伤均可诱导线粒体选择性自噬的发生,以减少铜毒性及抑制线粒体介导的细胞凋亡发生。线粒体为铜死亡发生的核心,线粒体选择性自噬极可能抑制铜死亡的发生,而自噬与铜死亡的关系仍需要进一步研究证实(图2)。
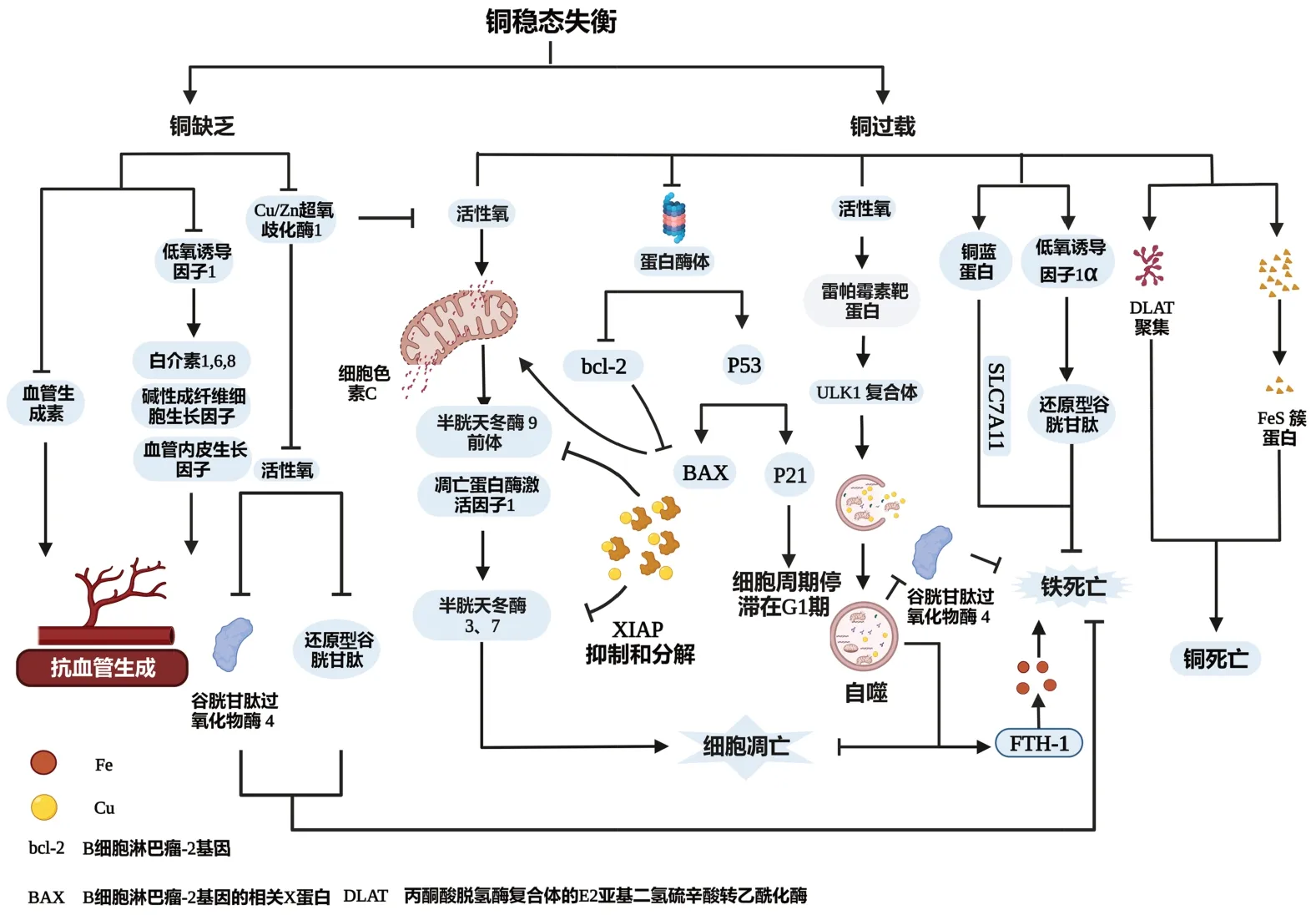
图2 铜缺乏或超载促进细胞损伤的机制示意图Figure 2 Schematic illustration of the mechanism by which copper deficiency or overload promotes cell damage
2 铜稳态失衡与肝病的关系
2.1 铜与WD WD是一种常染色体隐性遗传病,由铜转运P型ATP酶(ATP7B)先天突变导致铜在肝脏及其他组织中累积[2],进而引发一系列病变。
近年来研究发现WD患者过量肝铜通过抑制核受体功能、导致线粒体功能障碍等引发WD的肝脏病变。核受体参与调节细胞基因表达,WD患者异常升高的铜通过抑制核受体进而影响细胞脂质代谢。肝脏X受体(liver X receptor,LXR)是WD早期铜作用的主要靶点之一,Hamilton等[23]单独使用LXR激动剂T0901317治疗ATP7B缺陷小鼠,结果显示肝纤维化和炎性细胞因子显著降低,肝功能和组织学得到改善。以往观点认为WD患者异常升高的肝铜主要通过诱发氧化应激导致线粒体功能障碍,但近年来研究发现在肝铜介导的氧化应激发生之前,线粒体铜较正常水平升高约200倍,引起线粒体内外膜交联、线粒体膜电位变化、ATP产生减少和抑制线粒体DNA复制[22,24-25],从而导致线粒体结构、功能障碍和细胞凋亡的发生。
更早诊断及开始铜螯合治疗可以明显改善WD患者预后及预防神经系统症状的发生,现有的诊断方法早期诊断WD比较困难,这就要求寻找新的诊断方法及生物标志物。近年来有研究发现血液相对可交换铜和ATP7B肽浓度在诊断WD中具有高敏感性和特异性,但实际应用需要进一步验证。
口服铜螯合剂和锌是WD患者一线治疗方法,长期口服的不良反应、神经系统症状恶化仍是WD治疗所面临的挑战,而肝细胞及线粒体导向铜螯合剂的研究有望改善这一困境。恢复ATP7B定位、功能的靶向分子疗法及基因治疗也在积极研发中[2,25]。铜可通过多种途径促进肿瘤的发生发展,但有证据表明WD患者肝脏恶性肿瘤发病率远低于其他病因[26-27],研究人员推测这可能与WD患者长期口服铜螯合剂掩盖了肝铜的“自然”分布相关[26]。但也有证据表明过量铜摄入可以抑制大鼠化学毒物诱导细胞癌变,而长期口服铜螯合剂导致肝铁积聚和随后对肝细胞的伤害,可能是长期口服铜螯合剂WD患者发生HCC的一个原因。总之,HCC是WD患者的一种罕见并发症,阐述这种关联有助于WD患者肝脏恶性肿瘤的早期诊断和管理。
2.2 铜与非酒精性脂肪性肝病(NAFLD) NAFLD是一组以肝脏中脂肪堆积为特征的疾病,包括非酒精性脂肪肝(non-alcoholic fatty liver,NAFL)和非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH),最终可导致肝硬化和HCC,流行病学调查显示全球NAFLD患病率约为25%,已成为最常见的慢性肝病[28]。有证据表明,铜稳态失衡与NAFLD的发病密切相关,既往研究[29]发现NAFLD动物模型的肝铜水平降低,同时成人和儿童NAFLD患者的血清、肝脏和头发中的铜水平均较对照组降低。Tosco等[30]对铜缺乏大鼠的研究强调胞内铜水平降低通过损伤细胞线粒体形态和功能,抑制游离脂肪酸β氧化,促进肝细胞脂肪变性。果糖通过抑制十二指肠CTR-1表达减少铜的吸收,Song等[31]实验发现高果糖饮食小鼠的血清铜及肝铜水平降低,而低铜会显著抑制细胞脂肪酸β氧化限速酶肉碱棕榈酰转移酶及上调细胞脂肪酸合成关键酶脂肪酸合成酶(fatty acid synthase,FAS)的表达,诱发小鼠肝脏脂肪变性及肝细胞损伤。
研究[32]表明,铜摄入不足可能与NAFLD有关。Tallino等[33]发现低铜和高蔗糖饮食会明显上调与肝脏炎症、纤维生成和FAS相关的基因转录和表达,在没有严重肥胖的情况下,诱导肝脏脂肪变性和损伤的发生。肝铜降低通过抑制CP的活性和肝脏铁转运蛋白的表达减少肝铁输出,Aigner等[32]通过肝脏活检显示NAFLD患者的肝铁含量增加,而铁超负荷也通过诱导氧化应激、胰岛素抵抗和肝脏炎症来促进NAFLD发展,这些因素在NAFLD的发生和发展中起着关键作用。
与上述结果相矛盾的是,许多肝病的流行病学特征、疾病进展和治疗策略存在性别相关差异[34]。最近的一项研究也表明,血铜水平与NAFLD之间的关系仅在男性患者中显著[35],这可能与雌性激素的保护作用及铜摄入量存在性别差异相关。Stättermayer等[36]发现仅在无代谢综合征的NAFLD患者中,肝铜水平和肝脂肪变性程度之间存在负相关,这一结果与Lan等[35]的发现相反,这种差异可能是作者在实验中未考虑性别因素。此外,Aigner等[32]发现相比NAFL,NASH患者的肝铜含量更低,这表明肝铜水平与NAFL/NASH的进展相关。然而,也有证据表明NAFLD进展至肝硬化阶段,肝铜水平明显升高,具体原因尚未可知[37]。这些矛盾的结果提示铜与NAFLD之间具有更为复杂的联系,在未来需要进一步研究阐述铜在NAFLD发生、发展中的作用。
2.3 铜与HCC HCC是肝脏最常见的恶性肿瘤,是世界肿瘤相关性死亡第四大原因[38]。有证据表明HCC患者血清及肝铜水平升高[39],而这种现象可能与HCC细胞ATP7B下调或大型胞饮作用密切相关,同时Davis等[40]也发现敲除HCC细胞CTR-1基因可明显抑制肿瘤细胞增殖和转移。研究表明铜可通过多种机制促进肿瘤形成,具体机制尚未完全阐明,但研究人员使用铜处理HCC细胞显示可增强其增殖和迁移能力。大多数HCC细胞MYC明显过表达,2018年Porcu等[41]发现HepaRG细胞中过表达的MYC和MYC关联因子X结合后,与CTR-1基因启动子结合并诱导其转录,通过提高胞内铜水平促进肿瘤细胞增殖和迁移。
LOX家族是铜依赖性赖氨酰氧化酶,人HCC组织中LOX表达水平明显升高,研究表明LOX家族通过TGF-β介导的P38MAPK-VEGF信号转导促进血管形成,通过H1F-1/LOX信号转导通路使得细胞外基质胶原蛋白相互交联,促进HCC细胞生长与转移[42]。同时HCC组织中LOXL-2水平与HCC患者总体生存期和疾病特异性生存期呈负相关[43]。四硫钼酸盐(tetrathiomolybdate,TTM)已被证明可通过影响LOX活性来抑制细胞增殖和头颈部鳞状细胞癌的骨破坏行为,但其在HCC中的应用未来仍需进一步研究[44]。
COMMD蛋白家族在铜代谢中起关键作用,COMMD1缺乏引起细胞ATP7B从胞质囊泡到胞质膜运输缺陷,肝铜排出受阻导致肝内铜水平异常升高[45]。2022年Yang等[20]研究发现COMMOD10通过改变胞内Cu/Fe平衡和HIF-1/CP通路来影响HCC细胞放射抵抗性和铁死亡的发生,因此铜螯合剂TEPA可作为HCC患者的潜在放疗致敏剂。
HCC细胞中异常铜稳态为其诊断和治疗提供了一个新的方向[40],Yoshii等[46]研究发现曲恩汀(一种铜螯合剂)可通过抑制内皮细胞增殖和诱导细胞凋亡显著抑制HCC的生长和血管生成。HCC细胞内因糖酵解增强导致乳酸含量上升,进而通过乳酸/NF-κB/IL-8轴促进肿瘤血管生成,而这也与经导管肝动脉栓塞术/肝动脉灌注化疗栓塞术(transarterial embolization/transarterial chemoembolization,TAE/TACE)术后肿瘤快速复发相关。2020年Davis等[40]证实铜与HCC细胞在缺氧条件下代谢重编程密切相关,TTM螯合HCC胞内铜后可降低HCC糖酵解水平,进而抑制肿瘤细胞增殖活性,而TTM与TAE/TACE联合治疗HCC的疗效也尚待进一步研究。肝脏负责吸收和代谢含有金属的复合物,Rezaei等[47]发现Cu(Ⅱ)复合物通过上调HepG2细胞抑癌基因P53和凋亡基因(BAX和半胱天冬酶-8)的表达以及下调抗凋亡基因BCL-2的表达,诱导细胞发生凋亡,同时一些新型铜复合物也被开发用于HCC的光动力治疗。
一项来自我国广东的临床研究[48]发现,HCC患者血清铜水平与肝癌特异性生存期和总体生存期显著相关,这表明循环铜水平可能是HCC生存期的独立预测因子。与原发性HCC相反,转移HCC细胞铜摄取增加,因此64Cucl2-PET-CT可用于非侵入性评估位于生理铜摄取低区域的HCC的颅内转移和其他肝外转移,可用于肝移植患者术前评估,对改善转移性HCC患者的预后具有重要意义。此外放射性铜同位素也可作为放射性药物用于HCC肝外转移的治疗。
3 展望
最近的研究揭示了铜代谢的分子机制及其在人类疾病中的作用,铜死亡的发现为疾病机制的研究以及诊断和治疗方式的改进提供了新的思路和期望,铜螯合剂和铜补充剂也已被广泛用于铜代谢引起的疾病中。近年来铜代谢失调与肝脏疾病之间的关系已被充分证实,除了WD、NAFLD和HCC等肝脏疾病外,还有一些证据表明酒精摄入[49]和HCV感染[50]可能会影响人类的铜代谢调节,未来进一步探索铜代谢与慢性病毒性肝病和非病毒性肝病之间的关系,将为阐明这些疾病的发病机制和寻找新的治疗目标提供理论证据。
利益冲突声明:本文不存在任何利益冲突。
作者贡献声明:高沿航负责课题设计;高沿航和柳涛进行查阅文献并起草论文;高沿航、柳涛、刘雅丽和张飞宇参与文章撰写及修改,校阅论文。
猜你喜欢
中南民族大学学报(自然科学版)(2022年3期)2022-05-08
中国土壤与肥料(2021年5期)2021-12-02
海洋通报(2021年1期)2021-07-23
中国洗涤用品工业(2021年5期)2021-06-20
生物学通报(2021年4期)2021-03-16
世界科学技术-中医药现代化(2020年2期)2020-07-25
西南军医(2016年6期)2016-01-23
西南军医(2015年2期)2015-01-22
癌变·畸变·突变(2014年1期)2014-03-01
食品科学(2013年15期)2013-03-11
