
超高效液相色谱-串联质谱法检测牛可食性组织中氟佐隆残留
2023-10-25 12:32王亦琳陈超超王鹤佳
中国兽药杂志 2023年10期
王亦琳,叶 妮,尹 晖,陈超超,王鹤佳,孙 雷
(中国兽医药品监察所,北京 100081)
氟佐隆(Fluazuron, 化学式C20H10Cl2F5N3O3,CAS:86811-58-7)又称啶蜱脲,氟啶蜱脲或吡虫隆,属于苯甲酰脲衍生物,是一种昆虫发育抑制剂[1]。其在水中的溶解度小于0.02 mg/L,是一种亲脂性物质[2],化学结构式见图1。氟佐隆曾经作为农药杀虫剂广泛应用于水果、蔬菜等种植行业[3-4]。在畜牧业,氟佐隆是一种新型的抗寄生虫药[5-6],主要用于反刍动物,作为蜱、螨、壁虱等外寄生虫的杀虫药。氟佐隆的作用原理是能抑制组成昆虫表皮的主要成份几丁质的合成,通过抑制昆虫蜕皮和新表皮的形成,延缓其发育;或使昆虫表皮缺乏硬度,导致幼虫死亡或形成畸形蛹而死亡[7]。研究发现通过皮下注射单剂量给与奶牛氟佐隆后,氟佐隆在脂肪中的分布远高于肌肉、肾脏和肝脏[8-9]。氟佐隆在牛体内代谢较少,组织和粪便中的未代谢的原型药物占总残留量的90%[7]。氟佐隆主要的消除途径是经粪便排泄,其次是通过肾脏排泄,还有少量通过胆汁排泄。为防止在畜牧生产中滥用氟佐隆,造成相关动物性食品中氟佐隆的残留,危害人类健康,我国2019年发布的《食品安全国家标准 食品中兽药最大残留限量》(GB 31650-2019)[10]中规定了氟佐隆的最大残留限量(MRL):在牛肌肉中为200 μg/kg,在牛脂肪中为7000 μg/kg,在牛肝脏和牛肾脏中均为500 μg/kg。
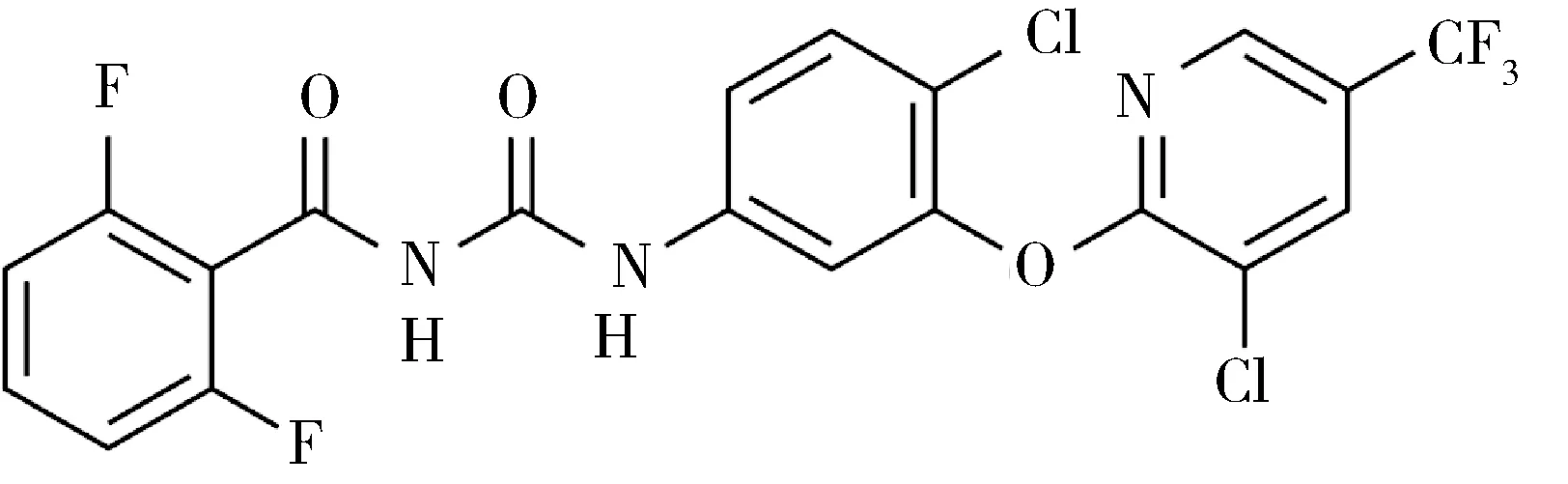
图1 氟佐隆化学结构式Fig 1 Chemical structure of fluazuron
目前国内外已经报道的氟佐隆的检测分析方法主要有气相色谱法[11],液相色谱法[12-14]和液相色谱-串联质谱法[15-17],但这些方法有的样品前处理过程比较繁琐,有的方法灵敏度不高,还有的分析基质为蔬菜等植物性食品,不能满足动物可食性组织中氟佐隆残留检测的实际需求。本文建立的超高效液相色谱-串联质谱法操作步骤简单,灵敏度高,重复性好,可以满足牛可食性组织中氟佐隆残留检测的要求,将对保障动物性食品安全,保证我国畜牧养殖业持续健康地发展发挥积极的作用。
1 材料与方法
1.1 仪器 日本岛津公司Shimadzu 30A超高效液相色谱仪-美国AB SCIEX公司QTrap 6500质谱仪(配电喷雾离子源); AE260电子天平,Mettler Toledo公司; Biofuge Strators高速冷冻离心机,贺利氏公司; SIR4涡旋混合器,IKA公司;固相萃取装置,Waters公司。
1.2 药品和试剂 氟佐隆对照品(C20H10Cl2F5N3O3,CAS:86811-58-7),品牌Bepure,购自北京振翔科技有限公司,含量99.5%。甲酸(色谱纯)、甲醇(色谱纯)、乙腈(色谱纯),美国Fisher公司;Captiva EMR-Lipid固相萃取柱:300 mg/3 mL,美国Agilent公司;所用水为超纯水。
1.3 标准溶液配制 精密称取氟佐隆对照品约10 mg,置于10 mL棕色量瓶中,用甲醇溶解并稀释成浓度为1 mg/mL的氟佐隆标准储备液。精密吸取1 mg/mL的氟佐隆标准储备液100 μL于10 mL量瓶中,用甲醇溶解并稀释成浓度为10 μg/mL的氟佐隆标准工作液。
1.4 样品前处理
1.4.1 样品的提取 称取试料各2±0.02 g于50 mL离心管中,加90%乙腈水溶液10 mL,加入一个陶瓷均质子,涡旋1 min,8000 r/ min离心5 min,得上清液备用。
1.4.2 样品的净化 取3 mL备用液过captiva EMR-Lipid小柱,挤干,收集流出液,过0.22 μm尼龙微孔滤膜,供液相色谱-串联质谱仪测定。
1.5 仪器条件
1.5.1 色谱条件 色谱柱为BEH C18色谱柱(50 mm×2.1 mm,粒径1.7 μm),流动相A相为0.1%甲酸水溶液,B相为0.1%甲酸乙腈溶液,梯度洗脱:0~0.2 min保持30% B;0.2~3 min,30% B线性变化到95% B;3~4 min 保持95% B;4~4.1 min 95% B线性变化到30% B;4.1~5.5 min 保持30% B。流速:0.5 mL/min;柱温:30 ℃;进样量:5 μL。
1.5.2 质谱条件 电喷雾离子源(ESI+);电喷雾电压为5500V;离子源温度为500 ℃;辅助气1为55 psi;辅助气2为55 psi;气帘气为30 psi;碰撞气为Medium;检测方式为多反应监测模式(MRM)。氟佐隆定性、定量离子对及对应的去簇电压、碰撞能量见表1。

表1 氟佐隆MRM参数Tab 1 Mass parameters of fluazuron
1.6 基质匹配标准曲线的制备 精密量取氟佐隆标准工作液适量,用空白试料经1.4提取净化后的流出液稀释成含药物浓度分别为1、2、5、10、40、80和100 ng/mL(相当于5、10、25、50、200、400和500 μg/kg添加浓度,针对牛肌肉)以及1、2、10、50、100、200和500 ng/mL(相当于5、10、50、250、500、1000和2500 μg/kg添加浓度,针对牛肝脏、肾脏和脂肪)的系列基质匹配标准工作液,过微孔滤膜后由低浓度到高浓度依次上机测定。以氟佐隆的特征离子质量色谱峰面积为纵坐标,氟佐隆基质匹配标准溶液浓度为横坐标,绘制标准曲线,并计算回归方程及相关系数。
1.7 方法灵敏度确定 添加适量浓度的氟佐隆标准工作液于2 g空白牛肌肉、肝脏、肾脏和脂肪中,经前处理后测定,观察药物特征离子质量色谱峰信噪比(S/N) 和对应药物浓度,以 S/N>3(按PtP算)作为方法的检测限,以 S/N>10作为方法的定量限。
1.8 方法准确度及精密度的测定 采用标准添加法,在空白牛肌肉、肝脏、肾脏和脂肪中添加定量限、MRL、2MRL三个浓度的氟佐隆标准工作液。在牛肌肉中的添加浓度为10、200、400 μg/kg;在牛肝脏和肾脏中的添加浓度为10、500、1000 μg/kg;在牛脂肪中的药物浓度为10、7000、14000 μg/kg。(由于牛脂肪的后两个添加浓度较高,为了保护质谱仪,避免样品过载污染仪器和后续样品,对MRL和2MRL这两个添加浓度点的脂肪样品进行稀释处理:样品按1.4项前处理后,将收集的流出液用空白脂肪样品经前处理得到的流出液稀释10倍后过滤,上机测定)每种浓度5个样品平行试验,重复3次,按照1.4项样品前处理方法处理之后上机测定,基质匹配标准溶液外标法定量,计算回收率、批内、批间变异系数。
1.9 基质效应的考察 取空白牛肌肉、肝脏、肾脏和脂肪样品,按1.4项进行样品前处理,用空白样品流出液配制成氟佐隆浓度为:牛肌肉2、40、80 ng/mL,牛肝脏和肾脏2、100、200 ng/mL,牛脂肪2、140、280 ng/mL的氟佐隆标准溶液,作为空白基质匹配标准溶液,每个浓度6份。用初始流动相配制成氟佐隆浓度分别与上述浓度相对应的标准工作液,作为基质效应考察的参照溶液。对上述样品进样分析,记录氟佐隆的峰面积并计算基质效应。
基质效应计算公式:基质效应%=基质匹配标准溶液的峰面积/参照溶液的峰面积×100%。
2 结果与分析
2.1 基质匹配标准曲线 以氟佐隆的特征离子质量色谱峰面积与其对应的基质匹配标准溶液浓度作图,得到相应的标准曲线,线性回归方程及决定系数(R2)见表2。氟佐隆在牛肌肉1~100 ng/mL的的基质匹配标准溶液浓度范围内;在牛肝脏、肾脏和脂肪1~500 ng/mL的基质匹配标准溶液浓度范围内,特征离子质量色谱峰面积与浓度均呈良好的线性关系,R2均>0.990。

表2 氟佐隆标准曲线的回归方程和决定系数Tab 2 Regression equations and coefficient of determination of fluazuron
2.2 方法灵敏度 按1.7项中所述方法进行处理,依据氟佐隆特征离子质量色谱峰信噪比S/N>3和S/N>10分别为方法的检测限和定量限,得出在空白牛肝脏、肾脏、肌肉和脂肪中,氟佐隆的检测限均为5 μg/kg,定量限均为10 μg/kg,特征离子质量色谱图见图2。

1. 牛肌肉;2. 牛肝脏;3. 牛肾脏;4. 牛脂肪1. Cattle muscle;2. Cattle liver;3. Cattle kidney;4. Cattle fat图2 10 μg/kg空白牛可食性组织添加样品中氟佐隆药物特征离子质量色谱图Fig 2 Characteristic ion mass chromatogram of fluazuron 10 μg/kg added in blank cattle edible tissues
2.3 方法准确度及精密度 在空白牛肌肉、肝脏、肾脏和脂肪中各添加3个不同浓度的氟佐隆标准工作液进行回收率试验,结果汇总见表3。试验结果表明,氟佐隆在牛肌肉10~400 μg/kg、牛肝脏和牛肾脏10~1000 μg/kg、牛脂肪10~14000 μg/kg空白添加浓度范围内的回收率为72.0%~98.6%;批内与批间RSD均小于15%。结果表明本方法定量准确,重复性好。
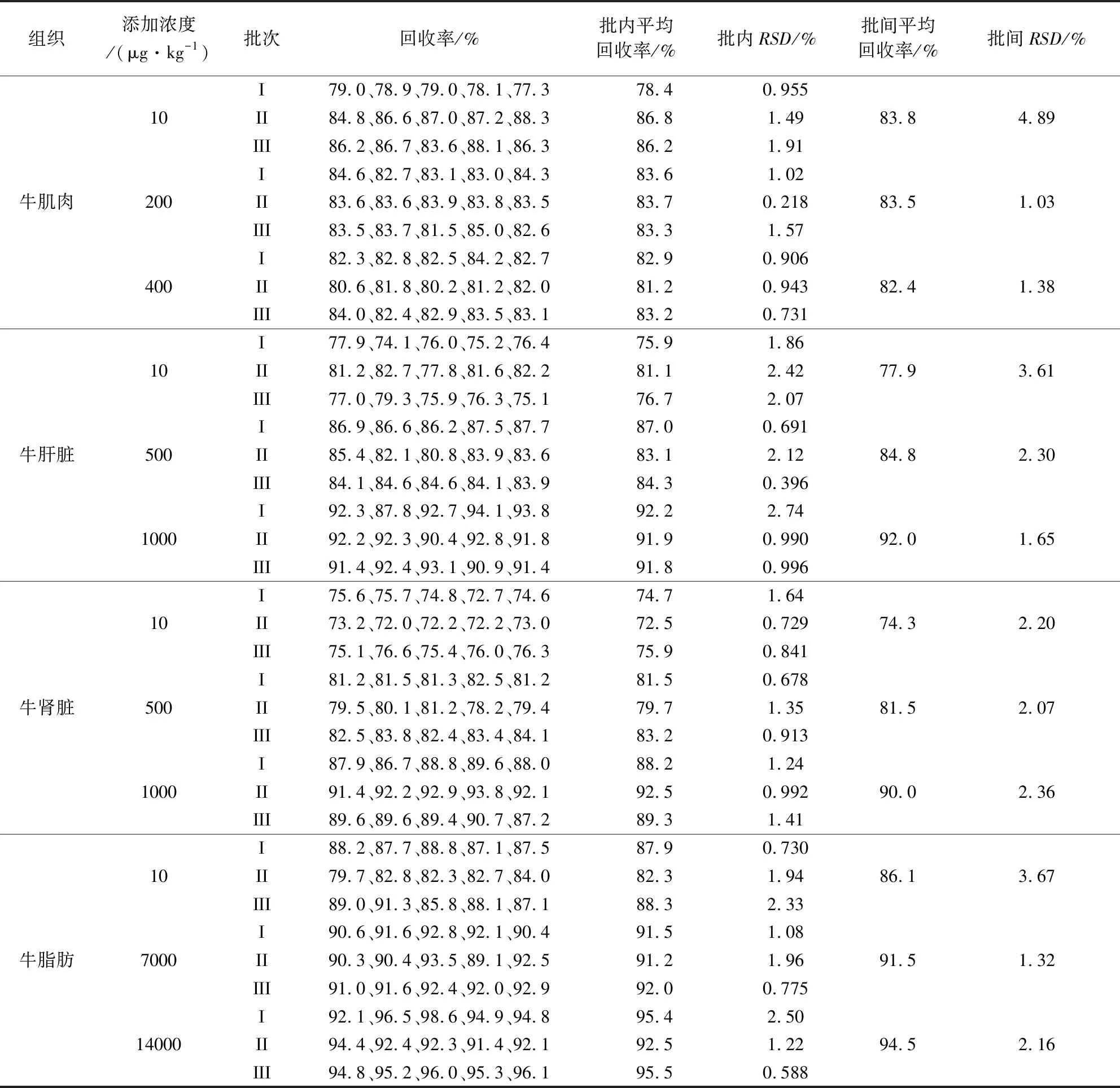
表3 空白牛可食性组织中氟佐隆添加的回收率(n=5)Tab 3 Recoveries of fluazuron in blank cattle edible tissues
2.4 基质效应考察结果 按1.9项所述方法进行基质效应考察,结果汇总见表4。从考察结果可以看出本分析方法的基质效应较微弱,但考虑到实际样品检测时可能会处理来源较为复杂的样品,基质效应可能会有变化。因此,在定量方法选择时仍然采用了基质匹配标准溶液法进行定量,消除可能存在的基质效应带来的影响,提高定量的准确性。
3 讨论与小结
3.1 提取方法的选择 由于氟佐隆不溶于水,目前已报道的文献中多采用纯乙腈[3,13]或正己烷[15]等有机溶剂作为氟佐隆的提取溶剂。考虑到乙腈和正己烷等有机溶剂具有一定的毒性,且本方法的分析样品为牛肉等生物样品,在加入纯有机溶剂涡旋提取时样品容易成团,影响提取效率。经过比较,选用了5倍称样体积的90%乙腈水溶液作为提取溶剂,涡旋提取一次,能有效提取样品中的氟佐隆药物残留。为防止加入提取液之后样品结块,本方法在每个样品管中加入一个带有切面的陶瓷匀质子,在涡旋提取操作时可以有效防止样品结块成团,提高了提取效率。
3.2 净化方法的选择 在关于动物可食性组织中氟佐隆残留检测的文献中,有报道采用Florisil 固相萃取小柱[8]和中性氧化铝小柱[15]净化处理样品。由于氟佐隆的酸度系数(pKa)为8.61±0.46,偏碱性,试验中曾选择了Oasis MCX小柱和Bond Elut Plexa PCX小柱进行净化效果的考察。结果发现MCX与PCX柱的样品前处理过程操作复杂,处理样品耗时较长,与文献报道的净化方法相比并无明显优势。本试验最终选择了EMR-Lipid柱进行样品的净化。提取液经高速离心后直接上样于EMR-Lipid小柱,收集样品流出液后直接过滤膜上机测定。整个净化步骤相比文献报道的净化方法操作简单,无需将固相萃取小柱预先活化,也省略了氮气吹干样品和样品复溶等步骤。从得到的谱图和回收率数据中可以看出,该方法在有效去除基质干扰的同时,样品回收率较高,可以满足氟佐隆残留检测的要求。
3.3 滤膜材质的考察 试验初期发现使用PTFE材质的滤膜,药物吸附严重,可以使药物响应值下降约2个数量级。通过比较PTFE、PES、GHP和尼龙等多种材质的滤膜,最终确定了尼龙材质的滤膜基本无药物吸附,最终选择0.22 μm的尼龙膜过滤样品。
目前国内外尚未检索到检测牛可食性组织中氟佐隆残留的超高效液相色谱-串联质谱分析方法,本方法的建立填补了国内外相关领域的空白。该方法快速、便捷,具有良好的可操作性和重现性,方法灵敏度、准确度和精密度均能满足兽药残留分析方面的要求,具有良好的应用前景。
猜你喜欢
中老年保健(2022年3期)2022-08-24
中国土壤与肥料(2021年5期)2021-12-12
中国土壤与肥料(2021年5期)2021-12-02
现代临床医学(2021年6期)2021-11-20
化工设计通讯(2020年10期)2020-09-17
北京园林(2020年4期)2020-01-18
中国生殖健康(2019年10期)2019-01-07
特别健康(2018年9期)2018-09-26
Cancer Biology & Medicine(2016年4期)2017-01-13
中国氯碱(2016年9期)2016-11-16
