
靶向调控肿瘤相关巨噬细胞的小分子药物研究进展
2023-10-24 10:52金佳明张庆云王均伟胡立宏
药学进展 2023年9期
金佳明,张庆云,王均伟,胡立宏
(南京中医药大学药学院,江苏 南京,210023)
近年来,随着分子生物学、肿瘤免疫学的迅速发展,以选择性增强患者免疫应答为重点的免疫治疗已成为一种新的肿瘤治疗手段[1]。目前,无论是久热不止的细胞程序性死亡受体-1(programmed cell death 1,PD-1)、细胞程序性死亡-配体1(programmed cell death ligand 1,PD-L1)和细胞毒性T淋巴细胞相关蛋白-4(cytotoxic T-lymphocyte-associated protein 4,CTLA4)单抗,还是淋巴细胞激活基因-3(lymphocyte activation gene,LAG3)、肿瘤坏死因子受体超家族成员4(tumor necrosis factor receptor superfamily member 4,TNFRSF4)等靶点,无一例外都是围绕如何恢复T淋巴细胞功能或如何提高获得性免疫系统的功能进行研究[2]。然而,大部分实体瘤微环境中浸润的T细胞较少,导致患者对调控T细胞功能的免疫治疗药物不敏感[3]。事实上在整个肿瘤浸润区域,肿瘤相关巨噬细胞(tumorassociated macrophages,TAMs)才是肿瘤微环境(tumor microenvironment,TME)中最为丰富的免疫细胞,约占肿瘤间质免疫细胞总数的50%以上,与肿瘤的发生发展,尤其是免疫逃逸密切相关[4]。因此,除了围绕T细胞的免疫检查点抑制剂疗法,基于TAMs的免疫疗法也受到越来越多的关注[5]。相关抗体的研发已经取得了重大进展,给肿瘤患者带来了新希望。但是目前看来,抗体治疗存在价格高、不良反应大、无法口服等缺点,这就给靶向调控TAMs的小分子药物的开发提出了迫切的需求[6]。本文对调控TAMs的相关通路及其小分子抑制剂的研究进展进行了综述,以期为靶向调控TAMs的小分子药物的开发提供思考。
1 肿瘤相关巨噬细胞的功能概况
TAMs是在肿瘤组织中浸润的巨噬细胞,主要由单核细胞分化而来。肿瘤细胞分泌的集落刺激因子-1(macrophage-colony stimulating factor,CSF-1)、趋化因子配体2(chemokine ligand 2,CCL2)等因子能募集外周循环血中的单核细胞到TME中,继而单核细胞分化成巨噬细胞[7]。巨噬细胞是先天性免疫的重要组成部分,是一种“专业”的吞噬细胞,在人体内可以通过吞噬衰老和死亡细胞起到人体清道夫的作用。因此,最初认为TAMs是抗肿瘤效应细胞,能杀伤肿瘤细胞或呈递肿瘤抗原诱导机体免疫应答从而清除肿瘤,但随着研究的深入,人们发现TAMs的主要作用竟然是促肿瘤和免疫抑制等[8]。
巨噬细胞在不同微环境和刺激因子的作用下可以向不同的方向极化。根据活化状态、功能及分泌细胞因子的不同,巨噬细胞主要可分为经典活化的M1型巨噬细胞(促炎)和替代活化的M2型巨噬细胞(抗炎)[9],如图1所示。M1型巨噬细胞介导宿主对多种细菌、病毒和原生动物等微生物的防御,并在抗肿瘤免疫中发挥作用。它由白介素-12(interleukin 12,IL-12)、干扰素γ(interferon γ,IFN γ)、脂多糖(lipopolysaccharide,LPS)、Toll样受体(toll-like receptors,TLR)等因子诱导激活。M1型TAMs可分泌IL-1β、肿瘤坏死因子(tumour necrosis factor α,TNF-α)等促炎因子并介导活性氧(reactive oxygen species,ROS)与一氧化氮(NO)的释放,促进炎症反应,发挥抗肿瘤作用[10]。
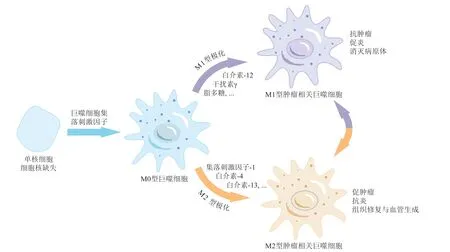
图1 TAMs的2种活化途径以及不同亚型TAMs的功能Figure 1 Two activation pathways of TAMs and the functions of different subtypes of TAMs
M2型巨噬细胞介导组织修复,促进肿瘤发生发展,它由IL-4、IL-13、CSF-1、转化生长因子-β(transforming growth factor-β,TGF-β)等细胞因子激活。M2型TAMs具有多种促肿瘤作用:1)促进肿瘤转移。M2型TAMs可分泌基质金属蛋白酶(matrix metalloproteinase,MMPs)、丝氨酸蛋白酶等蛋白酶破坏细胞外基质,从而促进肿瘤细胞和肿瘤间质细胞的侵袭与迁移[11]。2)刺激血管生成。M2型TAMs分泌血管内皮生长因子(vascular endo thelial growth factor,VEGF)、血小板衍生生长因子(platelet-derived growth factor,PDGF)、环氧合酶-2(cyclooxygenase-2,COX-2)、IL-10、MMP等细胞因子,促进肿瘤组织中血管和淋巴管生成[12]。3)抑制T细胞免疫功能。M2型TAMs可直接与髓源性抑制细胞(myeloid-derived suppressor cells,MDSCs)相互作用,抑制T细胞介导的抗肿瘤反应,还可表达PD-L1,通过PD-1/PD-L1通路抑制T细胞活化,诱导T细胞凋亡,进而发生免疫逃逸[13]。尽管M1型TAMs具有抗肿瘤作用,但随着肿瘤进展,M1型TAMs会逐渐极化为促肿瘤的M2型[14]。
综上,TAMs在人类恶性肿瘤的发展、转移以及耐药性中起着重要驱动作用,几乎参与了肿瘤发生发展的各个环节。因此,靶向TME中的TAMs是有效的肿瘤免疫治疗策略。目前已经确证了多个信号通路在调控TAMs功能中发挥重要作用(见图2),针对这些信号通路已经开发了多种调控TAMs的策略用于癌症的治疗[15]:1)抑制TAMs募集;2)减少TAMs数目;3)将M2型TAMs重塑为M1型;4)调节TAMs的吞噬作用。

图2 调控TAMs功能的信号通路以及调控策略Figure 2 Signaling pathways and regulatory strategies for regulating TAMs
2 调控肿瘤相关巨噬细胞功能的信号通路
2.1 趋化因子配体2/趋化因子受体2
CCL2是首个被发现的单核细胞趋化蛋白(monocyte chemotactic protein, MCP),可在多种细胞中产生(如肿瘤细胞、成纤维细胞、骨髓细胞)[16]。趋化因子受体2(chemokine receptor 2,CCR2)是一种G蛋白偶联受体,在单核细胞上表达[17]。肿瘤细胞过表达的CCL2与CCR2结合后可促使单核细胞募集到肿瘤部位,在肿瘤部位这些单核细胞进一步分化成TAMs诱导免疫抑制促进肿瘤生长[18]。CCL2/CCR2信号通路广泛参与多种恶性肿瘤的发生和进展(如胰腺癌、肾癌、肺癌),与患者的不良预后密切相关[19]。因此,靶向CCL2/CCR2有助于抑制TAMs募集,从而发挥抗肿瘤作用。
2.2 集落刺激因子1/集落刺激因子1 受体
CSF-1R是一种受体酪氨酸激酶,主要表达于单核细胞系,其受两种配体激活,即CSF-1以及IL-34[20]。CSF-1/CSF-1R信号通路可促进单核细胞,巨噬细胞等髓系细胞的生长、增殖与分化[21]。肿瘤细胞过表达的CSF-1与CSF-1R结合可促进人单核细胞分化为巨噬细胞,使TME中的TAMs浸润增加,促进TAMs向M2型分化激活。并且TAMs表面的CSF-1R与CSF-1结合后,可刺激TAMs分泌VEGF等多种细胞因子,进一步促进肿瘤的增殖、侵袭转移以及血管生成[22]。因此,阻断CSF-1R信号通路可抑制TAMs的增殖活化清除TAMs,促进M2型TAMs转变成M1型发挥抗肿瘤作用[23]。
2.3 整合素相关蛋白47/受体结合信号调节蛋白α
整合素相关蛋白47(cluster of Differentiation 47,CD47)是一种跨膜糖蛋白,其胞外区可作为SIPRα等配体[24]。SIRPα是SIRP家族的一种跨膜蛋白,主要表达于巨噬细胞、单核细胞等髓系细胞表面[25]。正常情况下,CD47可介导细胞增殖、迁移,T细胞活化,吞噬作用以及细胞凋亡。有助于维持正常细胞在生理条件下的免疫稳态,保护自身细胞不被误吞噬。然而,肿瘤细胞表面往往过表达CD47,其与巨噬细胞表面的SIRPα结合后可以引起SIRPα胞内的免疫受体酪氨酸的抑制性结构域(immunoreceptor tyrosine-based inhibition motif,ITIM)的酪氨酸磷酸化,然后募集并活化酪氨酸磷酸酶(src homology 2 domain-containing protein tyrosine phosphatase 1,SHP1)和SHP2,导致肌球蛋白IIA去磷酸化,抑制细胞的重构与收缩,从而阻止巨噬细胞的吞噬,导致肿瘤细胞的免疫逃逸[26]。因此,抑制CD47/SIRPα信号通路有助于重新激活巨噬细胞对肿瘤的吞噬作用,克服肿瘤细胞的免疫逃逸[27]。
2.4 细胞程序性死亡配体1/程序性细胞死亡受体1
PD-1是一种由CD28免疫球蛋白超家族的PDCD1基因编码的Ⅰ型跨膜蛋白,主要表达于T细胞、B细胞和巨噬细胞等免疫细胞。PD-L1是PD-1的主要配体,属于免疫球蛋白超家族B7成员[28]。通常认为PD-1/PD-L1通路通过抑制T细胞相关功能介导肿瘤免疫逃逸[29],但PD-1/PD-L1对TAMs的作用也不可忽视。研究发现,小鼠和人类的TAMs上都表达PD-1,并且表达量会随肿瘤增长而增加,且表达PD-1的TAMs往往是促肿瘤的M2型。TAMs的PD-1表达量与肿瘤细胞的吞噬效力呈负相关,因此,阻断PD-1/PD-L1通路会增强巨噬细胞对肿瘤细胞的吞噬作用,抑制肿瘤生长[30]。
2.5 主要组织相容性复合物I类/白细胞免疫球样蛋白受体B1
MHC-I是一种抗原呈递分子,是机体用于区分“自身”或“非自身”的标志物[31]。人类细胞中MHC是指人类白细胞抗原(human leukocyte antigen,HLA)[32]。通常认为,癌细胞表面的MHC-I可被细胞毒性T细胞识别从而杀死癌细胞[33]。最新研究表明,MHC-I在控制巨噬细胞吞噬中也发挥着核心作用,其可通过β2M亚基与巨噬细胞上的白细胞免疫球样蛋白受体B1(leukocyte immunoglobulin-like receptor B1,LILRB1)相互作用,产生吞噬抵抗作用,从而导致免疫逃逸。因此,阻断MHC-I与LILRB1的结合可增强巨噬细胞对肿瘤的吞噬作用[34-35]。因此,开发阻断MHC-I/LILRB1的药物可用来克服肿瘤的巨噬细胞吞噬逃逸作用[36]。
2.6 分化簇24/唾液酸结合免疫球蛋白Ig样凝集素-10
CD24是一种细胞黏附因子,主要参与细胞的识别、活化、信号转导等多种活动,其在多种肿瘤上高表达[37-38]。Siglec-10是一种抑制性受体,在单核细胞、B细胞等免疫细胞上表达,参与机体免疫调节。研究发现CD24是TAMs上高表达的Siglec-10分子的天然配体[39]。阻断CD24/Siglec-10相互作用可显著增强巨噬细胞对肿瘤的吞噬功能[40]。因此,CD24/Siglec-10也是一个巨噬细胞免疫检查点。
3 靶向调控肿瘤相关巨噬细胞小分子抑制剂的研究进展
上述信号通路在调节TAMs功能中发挥关键作用,与肿瘤发生发展及免疫逃逸密切相关。目前调控这些通路的主要手段为单克隆抗体以及小分子抑制剂,然而部分通路仅存在单克隆抗体而暂未发现有效的小分子抑制剂。其中已报道的部分小分子抑制剂已批准上市或进入临床研究阶段(见表1),本部分将根据调控TAMs的信号通路对代表性的小分子抑制剂进行总结。

表1 靶向调控TAMs小分子抑制剂的临床概况Table 1 Clinical overview of small molecule inhibitors targeting TAMs
3.1 集落刺激因子1受体小分子抑制剂
CSF-1/CSF-1R信号通路小分子抑制剂的研究是众多调控TAMs的信号通路中最广泛的,已经有多个抑制剂进入临床研究,其中PLX-3397已经获批上市。CSF-1R小分子抑制剂结合于CSF-1R蛋白的ATP催化结构域,由于该结构域在一些激酶中较为保守,所以大部分CSF-1R小分子抑制剂存在选择性不高的问题[41]。虽然CSF-1R与其同源的FMS样的酪氨酸激酶3(Fms-like tyrosine kinase,FLT3)、干细胞生长因子受体(c-kit receptor,KIT)等蛋白的结合口袋高度相似,但通过蛋白氨基酸序列叠合可发现在CSF-1R的Gly795位置,FLT3激酶为Cys828,KIT激酶为Cys809(见图3),Cys残基比Gly具有较大的空间位阻。因此,该区域是几种同源激酶的差异区域,有望利用该处关键差异设计出选择性的CSF-1R抑制剂[42-44]。
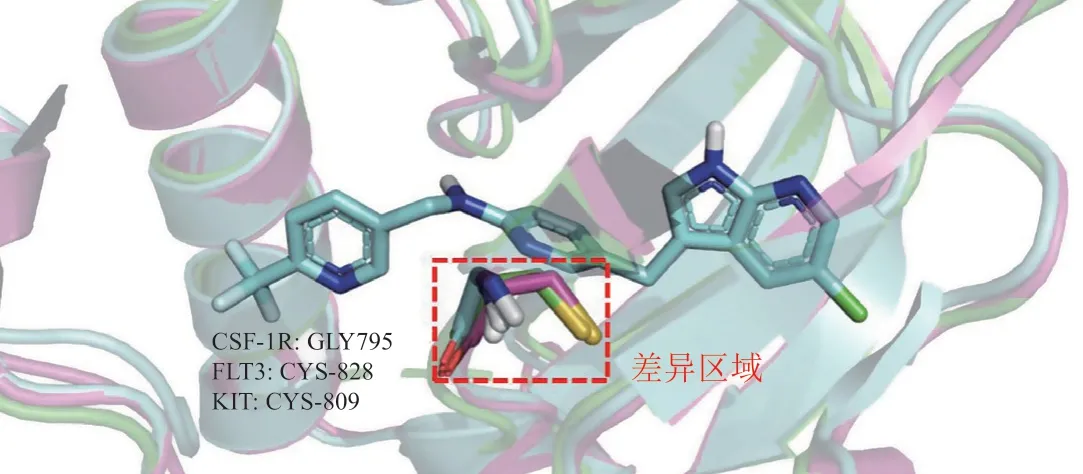
图3 CSF-1R与同源激酶的对齐图Figure 3 Alignment diagram of CSF-1R with homolo-gous kinase
3.1.1 氮杂吲哚类核心骨架氮杂吲哚(1)是通过高通量筛选获得,在化合物1的吡咯环上引入甲氧基取代的苄基得到化合物PLX-070(2),并在其基础上开发得到了PLX-647(3)、Pexidartinib(PLX-3397,4)等一系列化合物[45]。Pexidartinib是一种氮杂吲哚类非选择性的CSF-1R抑制剂(IC50= 20 nmol · L-1),于2019年获得美国FDA批准上市用于腱鞘巨细胞瘤的治疗。Pexidartinib的共晶结构(PDB ID:4R7H)显示,Pexidartinib通过氮杂吲哚上2个N原子分别作为氢键供体/受体与CSF-1R铰链区的氨基酸残基Glu664以及Cys666形成关键氢键作用,吡啶环上的氨基与Asp796形成氢键作用,三氟甲基取代的吡啶环伸入疏水口袋(见图4A)。但是该类化合物对其他激酶选择性不佳,易出现不良反应[46]。
Spangenberg等[47]通过在Pexidartinib吡啶环上引入F原子,并优化末端的疏水基团,得到了化合物PLX-5622,其CSF-1R抑制活性IC50为16 nmol · L-1,对同源的KIT以及FLT3的选择性大于20倍,目前已进入Ⅰ期临床研究。共晶结构(PDB ID: 6N33)显示,PLX-5622中间吡啶环上的F原子位于CSF-1R的氨基酸残基Gly795附近,是其选择性提升的主要原因(见图4B),考虑到选择性提高并不显著,可以尝试用空间位阻更大的基团替换F原子来进一步提高选择性。
3.1.2 吡啶类苗头化合物6是由迪加里加制药公司(Deciphera)通过高通量筛选获得[48],对其右侧吡啶环上取代基进行构效关系研究得到了化合物7,进一步在其酰胺氮原子上引入甲基得到了DCC-3014(8)。DCC-3014是一类以二吡啶醚为核心骨架的高选择性CSF-1R抑制剂。与氮杂吲哚类化合物相比,DCC-3014对CSF-1R选择性更好,其CSF-1R抑制活性IC50为3 nmol · L-1,对其同源的KIT的IC50为1 600 nmol · L-1,选择性大于500倍。DCC-3014与CSF-1R共晶结构(PDB ID: 7MFC)于同源蛋白KIT和FLT3的叠加图显示,DCC-3014吡啶环上的甲基位于几个同源激酶蛋白的差异区域,该甲基可容纳于CSF-1R中,不影响抑制剂与CSF-1R的结合,而与其他同源激酶的Cys残基可产生明显的冲突(见图5)。因此,在中间吡啶环上引入甲基,显著提高了DCC-3014对CSF-1R的选择性[49]。目前DCC-3014已进入Ⅲ期临床试验阶段,用于腱鞘巨细胞瘤的治疗。
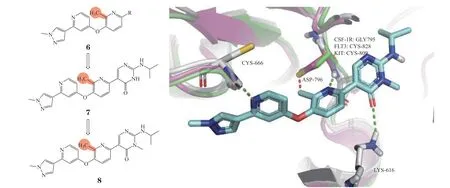
图5 DCC-3014与CSF-1R的共晶结构与同源蛋白对齐图Figure 5 Cocrystal structure and homeoprotein alignment of DCC-3014 with CSF-1R
3.1.3 苯并噻唑类BLZ945(9)是一种口服有效的苯并噻唑类选择性CSF-1R抑制剂(IC50= 1 nmol · L-1),对其同家族c-Kit、PDGFRβ、FLT3的选择性达到1 000倍以上[50]。BLZ945目前正在开展Ⅰ/Ⅱ期临床试验,作为单药或与斯帕巴他珠单抗(PD-1抗体)联用治疗晚期实体肿瘤。Czako等[51]基于BLZ945骨架进行改造设计合成了衍生物10,分子对接研究发现其苯并噻唑的2位处于CSF-1R的Gly795附近(见图6),在该位置引入甲氧基可提升与KIT 中Cys809残基的位阻,提高化合物11对CSF-1R的选择性。进一步在嘧啶氨基上引入吡唑环得到了化合物IACS-9439(12),其选择性显著优于BLZ945。

图6 化合物10与CSF-1R的分子对接图Figure 6 Molecular docking diagram of compound 10 with CSF-1R
3.1.4 氰基咪唑甲酰胺类化合物苗头化合物13由高通量筛选得到(IC50= 0.4 μmol · L-1),构效关系研究发现,在其杂环上引入吸电子基团(硝基、氰基等)对活性有利[52-54],在其苯环4位引入哌啶基团得到化合物14,其CSF-1R抑制活性和理化性质均得到改善。为避免苯胺结构的代谢毒性,将化合物7的6位哌啶环替换为环己烯基,得到化合物15与JNJ-28312141(16)。共晶结构显示,化合物15可结合于CSF-1R的DFG-inter构象(见图7A,PDB ID: 3KRJ)[55]。化合物17和化合物edicotinib(18)起源于对JNJ-28312141的选择性优化。Edicotinib是一种口服有效的氰基咪唑甲酰胺类选择性CSF-1R 抑制剂(IC50= 3.2 nmol · L-1),对其同源的KIT、FLT3的IC50分别为20和190 nmol · L-1[52],目前处于Ⅱ期临床试验阶段,用于急性髓细胞性白血病(acute myeloid leukemia,AML)的治疗。分子模拟研究发现该类结构中的二甲基环己烯基是其具有选择性的关键,化合物17的二甲基环己烯基可以延伸到激酶中含有Phe797的ATP-核糖区域,CSF-1R中的Phe797可以偏向小位阻的Gly795从而不与二甲基产生冲突,但KIT中的Phe811无法偏向大位阻的Cys807而与二甲基产生冲突(见图7B)[56],因此,Edicotinib 对CSF-1R的选择性较好。
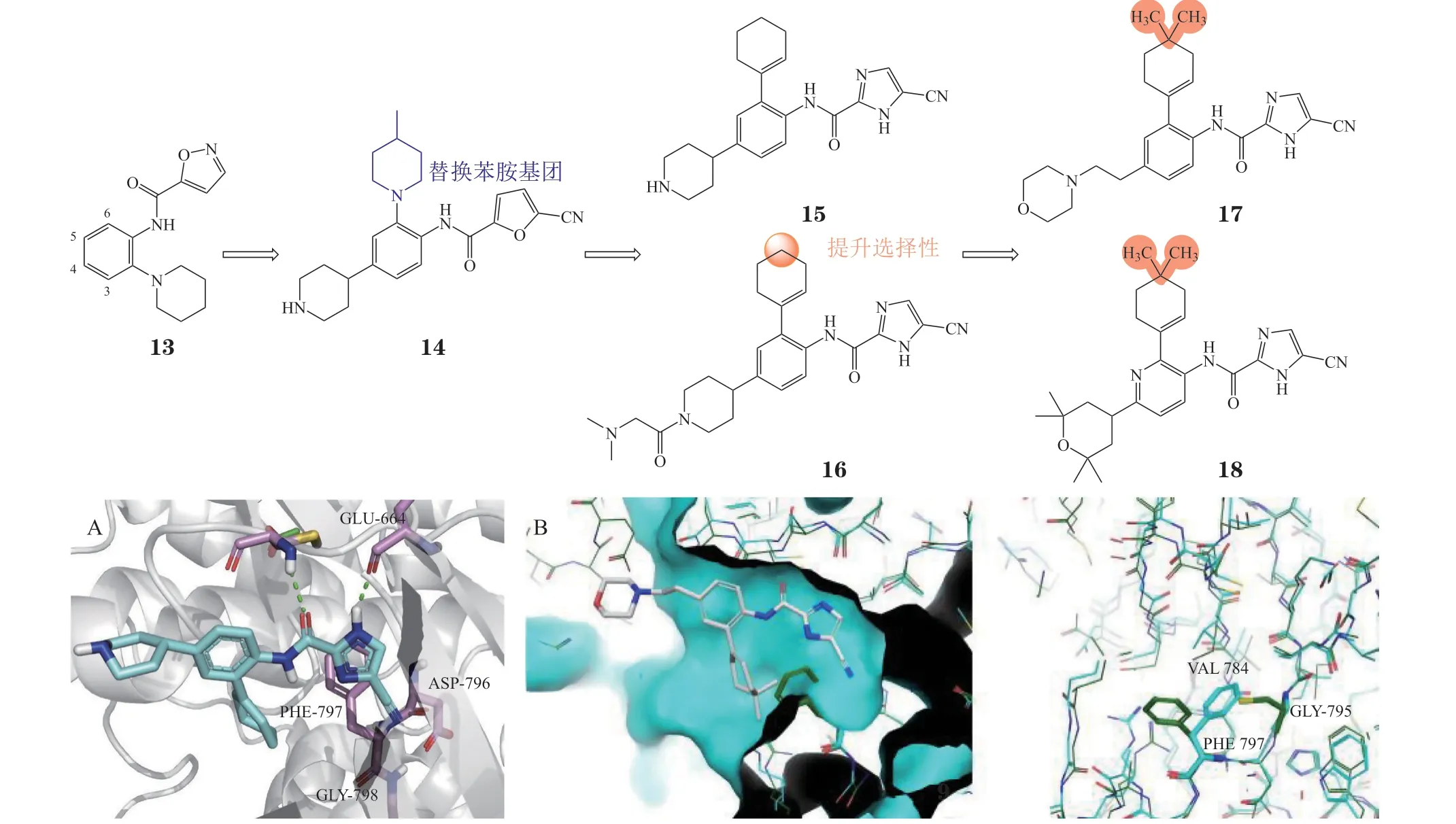
图7 化合物15与CSF-1R的结合模式以及化合物17与CSF-1R的分子对接图Figure 7 Binding mode of compound 15 with CSF-1R and molecular docking of compound 17 with CSF-1R
3.1.5 二氨基嘧啶类化合物GW-2580(19)是一种二氨基嘧啶类CSF-1R抑制剂(IC50= 60 nmol · L-1),具有较好的选择性[57]。分子对接研究发现,GW-2580的A环与B环通过亚甲基连接,呈L型交叉构象,A环与CSF-1R蛋白的铰链区结合,C环占据DFG-out状态下的疏水口袋,B环上的2个氧原子与Asp796形成双齿氢键,其甲氧基恰好被固定在Asp795附近,从结构上解释了其具有高选择性的原因。鉴于其高选择性,Japan Tobacco Inc.公司研发团队保留GW2580的B环与C环,引入氮杂环丁烷更好地固定A环与B环的交叉构象(见图8A),降低了小分子与CSF-1R结合时的能垒,并进一步在溶剂区引入乙二醇等亲水基团得到了活性更强的CSF-1R抑制剂JTE-952(20,IC50= 13 nmol · L-1)(见图8B)[58]。
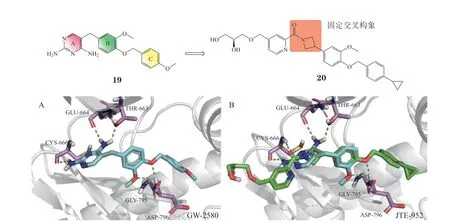
图8 GW-2580及JTE-952与CSF-1R的分子对接图Figure 8 Molecular docking of CSF-1R with GW-2580 and JTE-952
3.1.6 苯并咪唑类ARRY-382(21)是一种苯并咪唑类选择性CSF-1R抑制剂(IC50= 9 nmol · L-1),与PD-1单抗pembrolizumab联合治疗晚期实体肿瘤已进入Ⅱ期临床试验阶段[59],然而辉瑞公司最近发布的研究报告显示该方案对患者无有益效果,其临床研究也被终止。通过分子对接模拟其与CSF-1R蛋白的结合模式(PDB ID: 7MFC),推测其选择性是由于环丙基占据了口袋内的疏水空腔,导致苯并吡唑环靠近Gly795,而该位置可与其他同源激酶的半胱氨酸残基产生空间位阻,从而产生选择性(见图9)。
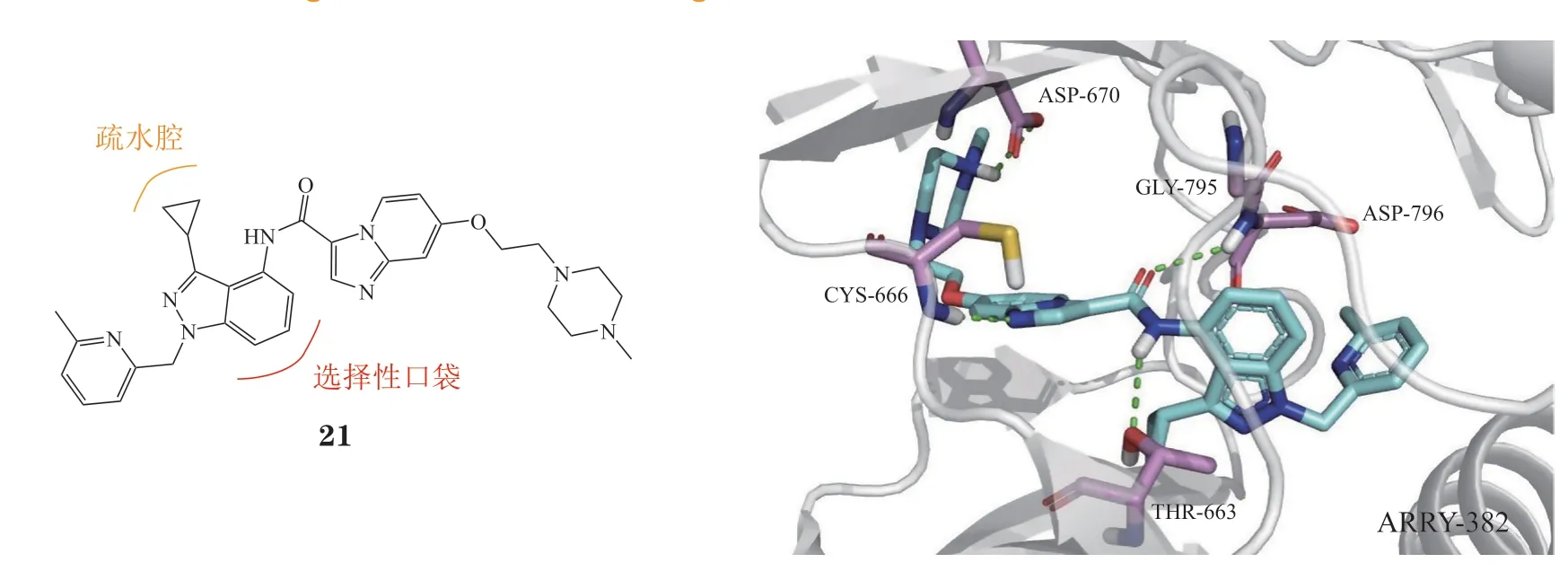
图9 ARRY-382与CSF-1R的分子对接图Figure 9 Molecular docking of CSF-1R with ARRY-382
3.2 趋化因子受体2小分子拮抗剂
CCR2小分子拮抗剂根据其结合位点可分为正构拮抗剂与变构拮抗剂。正构结合位点分为3个区域,即氢键结合区、脂质双层区以及芳环区(见图10A)。通过对比BMS-681、MK-0812 2种CCR2正构拮抗剂的晶体复合物可发现,2种分子在CCR2口袋内的空间走向大致相同,但互作残基却存在差异,BMS-681(22)的氨基吡咯烷酮与Tyr49、Thr292形成氢键,三氟甲基喹唑啉进入脂质双层区,而三取代环己烷则位于Trp98所在的芳环区(见图10B)[60];MK-0812(23)则通过其环己胺以及酰胺与Glu291、Tyr49形成氢键,三氟甲基芳基进入脂质双层区,而四氢吡喃位于Tyr120的所在的芳环区(见图10C)[61]。

图10 BMS-681以及MK-0812与CCR2正构位点的结合模式图Figure 10 Binding modes of BMS-681 and MK-0812 to CCR2 orthosteric site
代表性的变构拮抗剂为CCR2-RA-[R](24),其结合于CCR2的G蛋白结合位点附近,通过阻断CCR2激活构象的形成来非竞争性地拮抗CCR2。共晶结构显示(见图11),CCR2-RA-[R]的酰胺基与羟基分别作为氢键受体与Phe312和Lys311形成氢键相互作用,苯环与Tyr305形成pi-pi堆积相互作用,而环己基位于Val244等残基形成的疏水口袋中(PDB ID:5T1A)[60]。目前进入临床试验的CCR2拮抗剂主要为正构拮抗剂,下文主要介绍这些进入临床研究的CCR2正构拮抗剂的研发历程。

图11 CCR2-RA-[R]与CCR2变构位点的结合模式图Figure 11 Binding mode of CCR2-RA-[R] to CCR2 allosteric site
3.2.1 氨基吡咯烷类拮抗剂因塞特公司(Incyte)通过对已报道的CCR2拮抗剂25与26进行结构分析,发现它们的核心骨架由中心的叔胺以及两端亲脂性芳环构成,叔胺N原子与CCR2的Glu291形成的氢键为关键相互作用[62]。因此,该团队在叔胺N原子与芳环之间插入脂肪环以提升N原子电荷密度,增强氢键相互作用,成功发现了一种氨基吡咯烷类的CCR2拮抗剂INCB3344(27,IC50= 7.8 nmol · L-1)。然而INCB3344具有一定的hERG钾离子通道(human ether-a-go-go related gene potassium ion channel)抑制活性,会导致心脏毒性,还会抑制细胞色素P450 3A4酶(cytochrome P450 3A4,CYP3A4)。为解决该问题,该团队在INCB3344系列化合物中选择对hERG毒性较低的化合物28作为先导化合物,通过去掉吡咯烷环上的乙氧基得到了无明显hERG毒性的CCR2拮抗剂INCB3284(29,IC50= 3.7 nmol · L-1)[63](见图12)。
接下来,Incyte研发团队通过翻转氨基吡咯烷骨架发现了新一代CCR2拮抗剂30(IC50= 35 nmol · L-1),进一步围绕环己基4位取代基进行优化,得到化合物PF-04136309(31,IC50= 5.2 nmol · L-1)(见图13),其对人的CCR2选择性高、hERG毒性弱、且具有较好的吸收、分配、代谢、排泄和毒性(absorption,distribution,metabolism,excretion,and toxicity,ADMET)性质[64],目前正在开展与吉西他滨、白蛋白紫杉醇联合治疗胰腺癌的Ⅱ期临床试验。
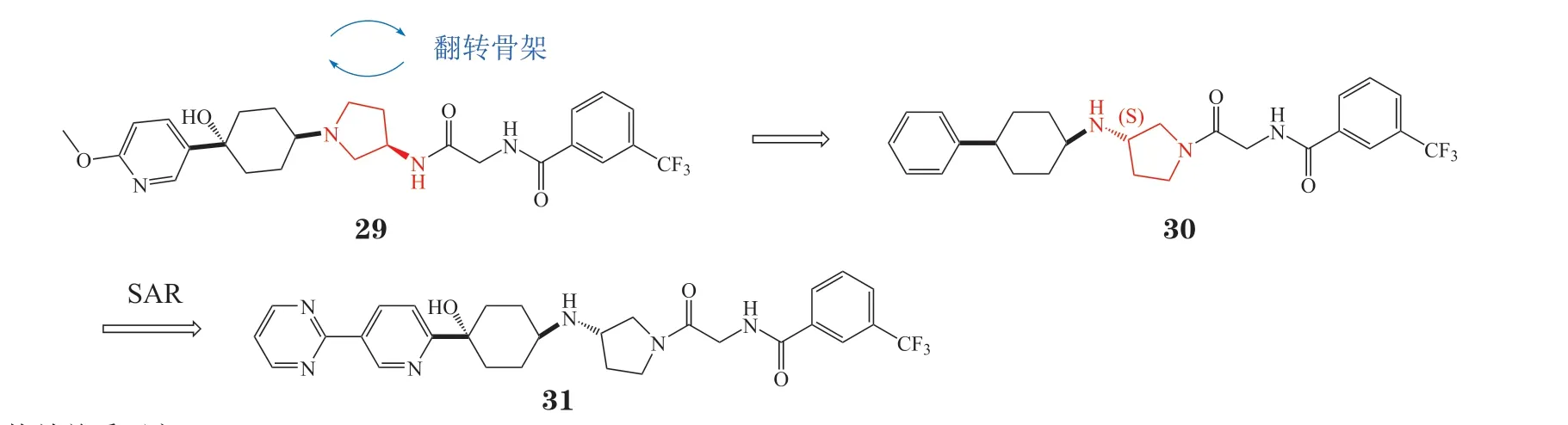
图13 PF04136309的研发历程Figure 13 Development process of PF04136309
3.2.2 三取代环己基类基于氨基吡咯烷类CCR2拮抗剂32,百时美施贵宝(BMS)公司研发团队在保持N原子空间位置不变的情况下用环己烷替代吡咯环,设计合成了化合物33[65],叠加图显示两者的N原子几乎处于同一位置。进一步研究发现顺式环己烷的化合物34活性更强,在三氟甲基的对位引入大基团时CCR2拮抗活性显著提升,其中化合物35活性最佳(IC50= 5.1 nmol · L-1)。为改善化合物35的成药性,研究团队利用砜替换酰胺键,去掉大体积的脲结构,并在环己基4位引入叔胺得到了活性更强的化合物36(IC50= 1.3 nmol · L-1)[66],其活性提升可能与三取代环己基构象更稳定有关(见图14)。

图14 BMS-813160的研发历程Figure 14 Development process of BMS-813160
为进一步提高三取代环己烷类CCR2拮抗剂的药代动力学性质,BMS研发团队在化合物36的基础上通过大量构效关系研究得了一种CCR2/5双靶点拮抗剂化合物37,其IC50分别为2.7和6.3 nmol · L-1。由于化合物37在人全血(human whole blood,HWB)中的CCR5拮抗活性较弱(IC50=34.7 nmol · L-1),在其右侧喹唑啉部分引入叔丁基吡唑环得到BMS-813160(38,CCR2/5 IC50为6.2和3.6 nmol · L-1),其在HWB中拮抗活性显著提升(HWB CCR2/5 IC50为4.8和5.7 nmol · L-1)。BMS-813160在生理pH下(pH = 7.4),叔丁胺会与乙酰胺的“NH”间形成分子内氢键,从而形成1,3-双直立构象,这种构象屏蔽了环己烷下方的极性基团从而改善了化合物的疏水常数(cLogP),提高了透膜性,有利于提高其口服生物利用度[67](见图14)。
3.3 整合素相关蛋白47/受体结合信号调节蛋白α小分子抑制剂
目前CD47的小分子抑制剂的主要作用机制可分为:直接阻断CD47与SIRPα结合;在转录和翻译水平抑制CD47表达;在翻译后修饰水平影响CD47与SIRPα的结合[68]。已经报道的一些单克隆抗体或多肽类药物可以直接结合于CD47或SIRPα蛋白,阻断两者结合,但目前很少有可直接结合CD47/SIRPα非肽类小分子抑制剂的报道。
3.3.1 直接阻断整合素相关蛋白47/受体结合信号调节蛋白α结合的小分子抑制剂1)NCGC00-138783 Burgess等[69]通过高通量筛选发现化合物NCG00138783(39)可有效抑制CD47与SIRPα的结合(IC50= 50 μmol · L-1),并且其衍生物NCGC00138419(40)和NCGC00138430(41)都可以抑制CD47/SIRPα的相互作用,表明该类结构有望作为一种全新母核用于CD47/SIRPα抑制剂的开发。有研究对NCGC00138783与CD47/SIRPα的结合模式进行了预测(见图15)[70],发现其比CD47更容易与SIRPα结合。NCGC00138783中的[1,2,4]三唑[1,5-c]喹唑啉位于疏水空腔,可与SIRPα的Phe74形成pi-pi堆积相互作用,与Gly34形成氢键,末端酰胺基可与SIRPα中的Gln52形成氢键,这些残基都是CD47/SIRPα相互作用中的关键残基。
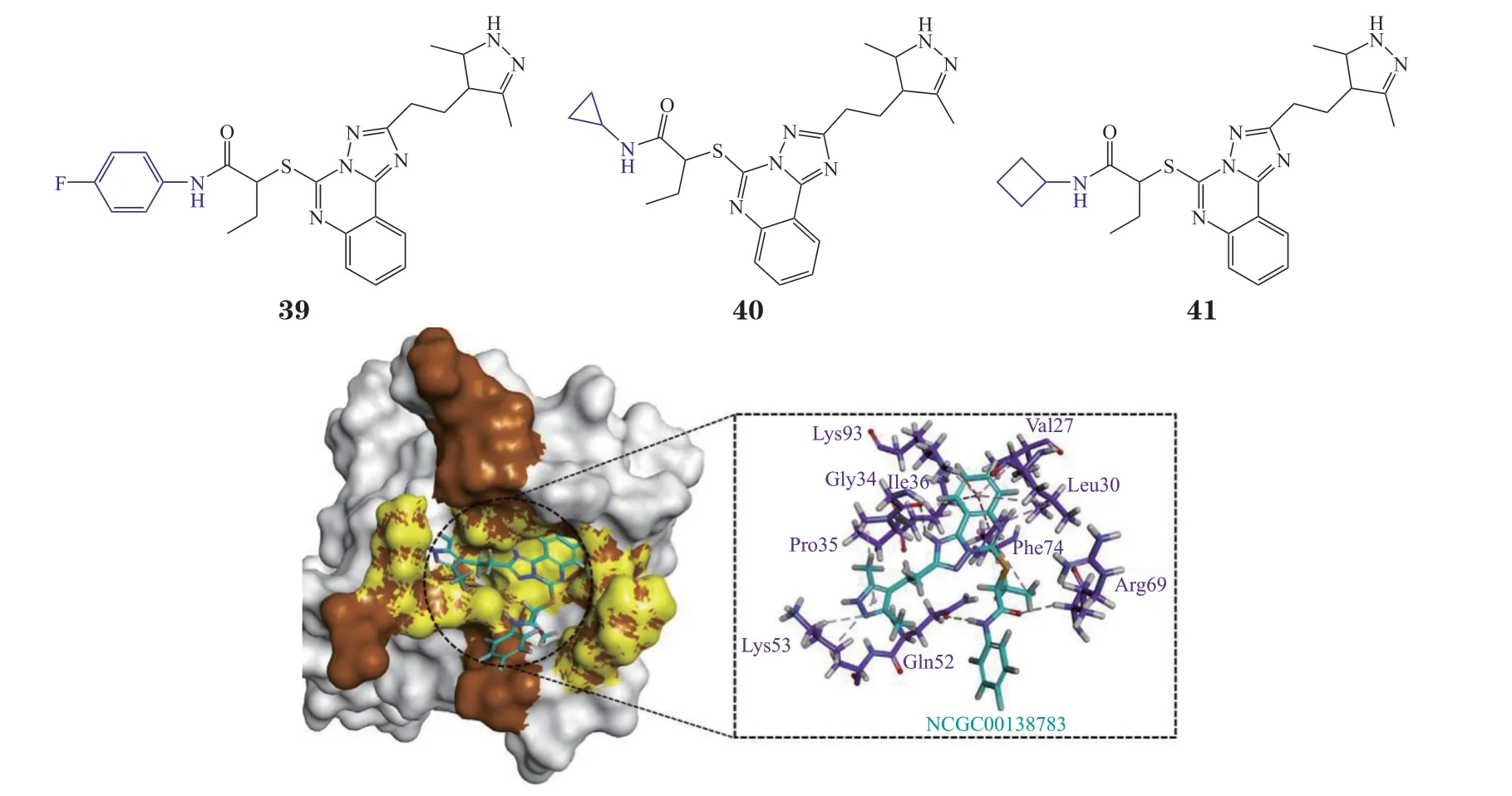
图15 NCG00-138783与SIRPα分子对接图Figure 15 Molecular docking of NCG00-138783 with SIRP-α
2)多肽类小分子抑制剂 Wang等[71]利用高通量筛选得到一种直接结合于CD47的小分子多肽Pep-20,其与人及大鼠CD47结合的Kd值分别为2.91和3.63 μmol · L-1。Pep-20能够显著增强巨噬细胞对多种肿瘤细胞的吞噬能力,且效果优于CD47单抗B6H12。Hu等[72]在Pep-20基础上进行结构简化,去掉其N端2个氨基酸,C端1个氨基酸,得到了多肽Pep-20(2-10),其与CD47的对接结果显示,CD47与SIRPα接触面的Phe4、Lys6、Glu104和Glu106是关键结合位点(见图16A),为靶向CD47抑制剂的开发提供了关键信息。Wang等[73]对CD47/SIRPα结合平面进行分析,发现残基Leu3、Lys6、Tyr37、Glu97、Glu104以及 Glu106对CD47与SIRPα结合的贡献较大,依据此设计了多肽RS17,通过分子模拟发现RS17的N端可与CD47的Lys6、 Glu104等氨基酸残基相互作用,C端可与Glu106、Leu3、Lys75、Asp77等残基相互作用(见图16B)。RS17与CD47结合的亲和力极强(Kd= 3.86 nmol · L-1),其可显著增强巨噬细胞吞噬肿瘤细胞的作用,且与CD47单克隆抗体具有相似的疗效。除了靶向CD47的多肽类抑制剂外,D4-2是一种靶向结合SIRPα的多肽(Kd= 10 nmol · L-1),其可与SIRPα的氨基酸残基Ala5、Asp84、Pro11、Phe51和Phe56相互作用,从而阻断SIRPα与CD47的结合[74]。

图16 Pep-20(2-10)以及RS17与CD47的分子对接图Figure 16 Molecular docking of Pep-20 (2-10) and RS17 with CD47
3.3.2 在转录和翻译水平抑制整合素相关蛋白47表达的小分子抑制剂1)RRx-001 RRx-001(42)原本被用于航天航空工业,后来被应用于抗肿瘤药物研究[75]。RRx-001可通过其结构中的α溴代酮作为亲电共价弹头与血红蛋白β的Cys93残基共价结合。结合了RRx-001的红细胞随血液循环进入肿瘤微环境,黏附在缺氧的肿瘤微血管上,然后被TAMs吞噬[76]。作用机制研究发现,RRx-001是通过抑制原癌基因MYC表达,下调CD47/SIRPα的表达发挥抗肿瘤作用(见图17)[77]。构效关系研究发现,将RRx-001共价弹头中的溴原子替换为氯原子或碘原子会导致其抗肿瘤活性降低,表明α溴代酮的共价弹头是其关键药效团[78]。

图17 RRX-001的起源以及作用机制Figure 17 Origin and mechanism of RRX-001
2)调控CD47表达的经典药物 很多作用于其他靶点的经典药物也被发现具有抑制CD47表达的功能。吉非替尼(gefitinib,43)是用于治疗非小细胞肺癌的第一代表皮生长因子受体酪氨酸激酶抑制剂,最近有研究发现,吉非替尼可显著下调非小细胞肺癌细胞中CD47的表达,增加巨噬细胞的吞噬作用[79]。二甲双胍(44)是临床上治疗Ⅱ型糖尿病的一线药物,其也具有抗炎抗癌等多种疗效,是一种公认的多靶点药物,有研究发现二甲双胍可下调乳腺癌细胞CD47 的表达,抑制乳腺癌肿瘤干细胞增殖并且提高巨噬细胞的吞噬能力,但是其具体机制尚不明确[80]。JQ-1(45)是一种溴结构域和超末端蛋白(bromodomain and extra-terminal,BET)小分子抑制剂,属于表观遗传学药物,其也可以通过抑制c-Myc原癌基因下调CD47的表达[81]。
3.3.3 在翻译后修饰水平影响整合素相关蛋白47/受体结合信号调节蛋白α结合的小分子抑制剂翻译后修饰(post-translational modifications ,PTMs)是指蛋白质在翻译后的化学修饰过程,可促使蛋白质成熟并发挥功能[82]。谷氨酰环化酶同工酶(glutaminyl-peptide cyclotransferase-like protein,QPCTL)是一种PTMs相关酶,属于谷氨酰环化酶(glutaminyl cyclases,QCs)的一种亚型,其可催化靶蛋白上N端谷氨酰胺和谷氨酸残基环化为焦谷氨酸残基[83]。QPCTL催化CD47与SIRPα结合平面上N端的焦谷氨酸残基的形成,该残基对CD47/SIRPα的结合十分重要。因此,抑制QPCTL的活性可以阻断CD47与SIRPα的结合,恢复巨噬细胞对肿瘤细胞的吞噬作用[84]。
由于CD47在红细胞上同样高表达,直接靶向CD47会导致贫血不良反应,而QPCTL在红细胞内不表达,因此,抑制QPCTL可避免该类不良事件的发生[85]。近年来已经公开了多种有效的QPCTL抑制剂,如PBD-150(46)、SEN177(47)以及PQ912(48)等[86],共晶结构显示(见图18),该类抑制剂可通过结构中的咪唑或苯并咪唑基团与QCs中的锌离子配位[87]。目前研究已证实QPCTL抑制剂能够阻断CD47/SIRPα的相互作用,促进巨噬细胞的吞噬,增强抗肿瘤作用[84]

图18 PBD-150及SEN177与谷氨酰环化酶的结合模式图Figure 18 Binding modes of QCs with PBD-150 and SEN177
3.4 细胞程序性死亡配体1/程序性细胞死亡受体1小分子抑制剂
以往研究中,PD-1/PD-L1抑制剂多用于调控T细胞功能,针对巨噬细胞的研究较少,但PD-1/PD-L1也是调控巨噬细胞的免疫检查点,因此,相关小分子抑制剂也可应用于调控巨噬细胞功能。目前已开发了一些肽类或间接调控PD-L1/PD-1通路的小分子抑制剂,但本文主要讨论可直接靶向结合PD-1/PD-L1的非肽类小分子抑制剂。目前直接靶向PD-1/PD-L1的抑制剂主要结合于PD-1/PD-L1蛋白互作平面,但该处为较平坦的疏水口袋,是设计小分子PD-1/PD-L1抑制剂需要面对的难题[88-89]。
3.4.1 间苯二酚二苄基醚类化合物BMS开发了一系列间苯二酚二苄基醚类PD-L1/PD-1抑制剂,其通式为49,该类化合物可有效阻断PD-1/PD-L1的相互作用,代表性的化合物BMS-202(50)和51的IC50分别为18 和22 nmol · L-1[90-91]。共晶结构(PDB ID:5J89)显示[92],化合物BMS-202可直接结合于PD-L1平坦的疏水空腔,使PD-L1形成二聚体,阻断PD-L1与PD-1的结合(见图19),其中PD-L1二聚体口袋内的多个酪氨酸残基(Tyr56、Tyr123)以及Met115与抑制剂形成的疏水作用十分关键。该研究首次解析了PD-L1/PD-1抑制剂与PD-L1的共晶结构,为后续抑制剂的开发奠定了结构生物学基础。
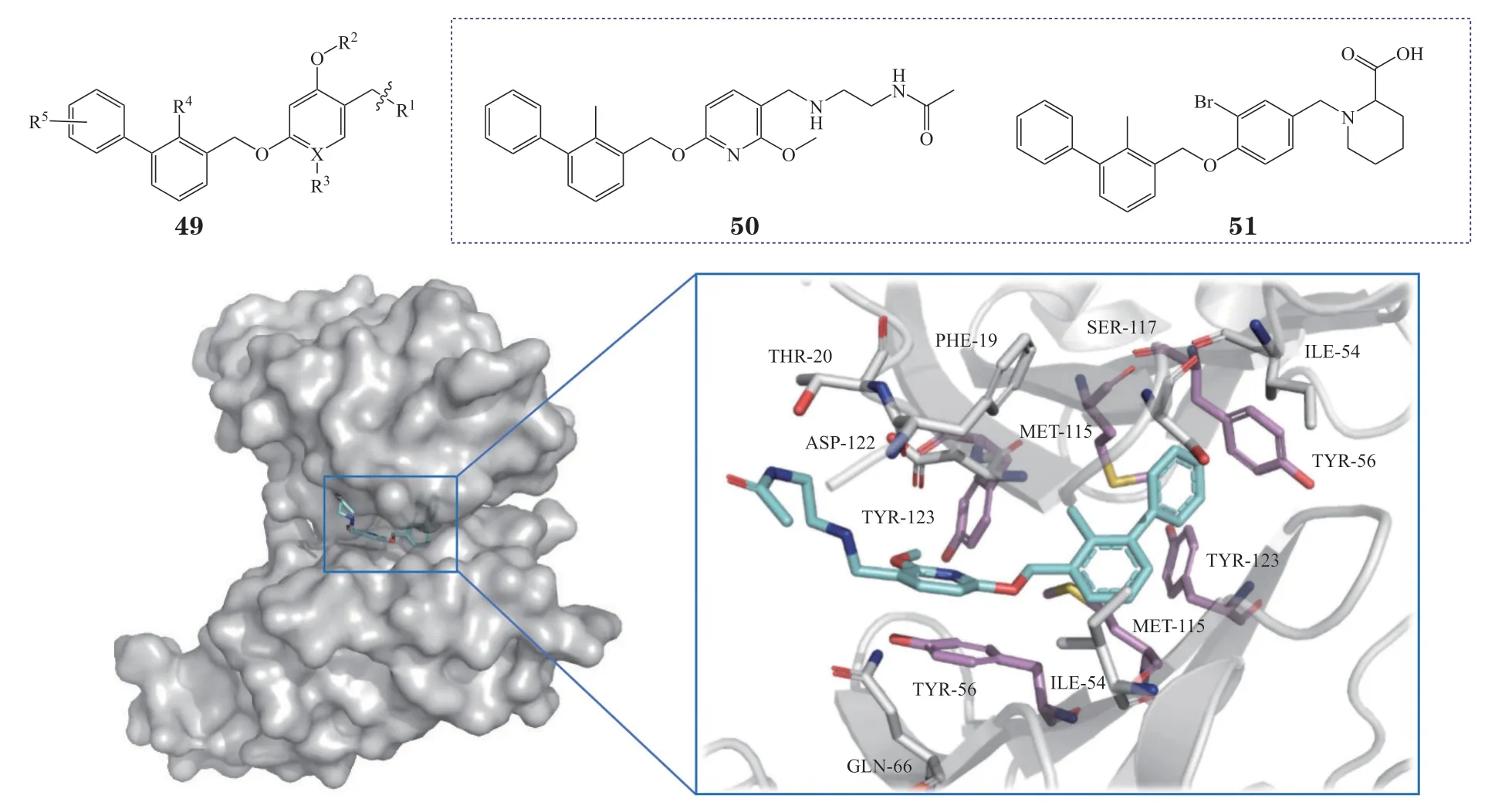
图19 BMS-202与PD-L1的结合模式图(PDB ID:5J89)Figure 19 Binding mode of BMS-202 with PD-L1
3.4.2 溴苄基醚类化合物基于BMS开发的间苯二酚二苄基醚类抑制剂,Wang等[93]以溴原子替代甲基得到了一系列溴苄基醚衍生物(见图20),该类化合物保持很强的PD-1/PD-L1抑制活性,其中化合物YPD-29B(52)的IC50达到了0.08 pmol · L-1。为提高化合物的透膜性,进一步对YPD-29B的羧基进行了叔丁酯前药修饰,得到前药IMMH-010(53)[94],成为我国首个获批临床试验的口服PD-L1小分子抑制剂,目前正在进行Ⅰ期临床试验。
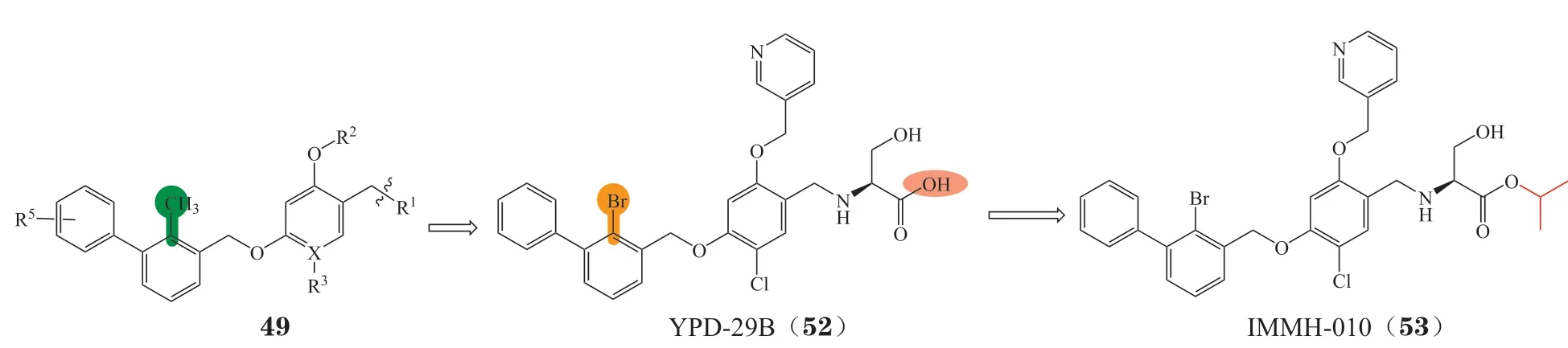
图20 IMMH-010的研发历程Figure 20 Development process of IMMH-010
3.4.3 芳基乙烯类化合物受间苯二酚二苄基醚类结构的启发,广州再极药业公司将该类化合物中的醚键用C=C键替代,得到一系列通式为54的芳香族烯烃类PD-1/PD-L1抑制剂[95]。分子模拟可发现其与BMS系列化合物的结合模式类似(见图21),代表性化合物55与56的IC50为18 和48 nmol · L-1,其中此类化合物MAX-10181(结构未公布)已在澳大利亚启动Ⅰ期临床试验。
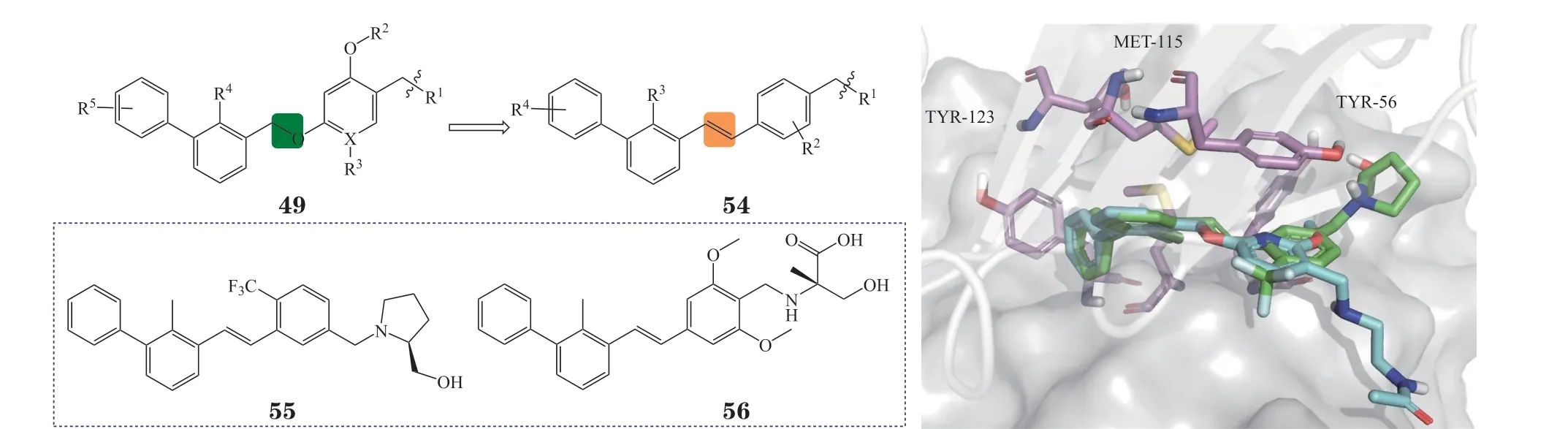
图21 化合物54与PD-L1的分子对接图Figure 21 Molecular docking of compound 54 with PD-L1
3.4.4 稠合杂环类化合物Incyte公司通过用稠合杂环替代间苯二酚二苄基醚类化合物中的苯环,设计合成了全新结构类型的小分子抑制剂[96],其中代表性的化合物为INCB086550(57),其对人PD-L1的IC50为3.1 nmol · L-1。INCB086550是首个口服的PD-1/PD-L1小分子抑制剂,目前已进入Ⅱ期临床试验(NCT04629339)用于实体瘤的治疗。

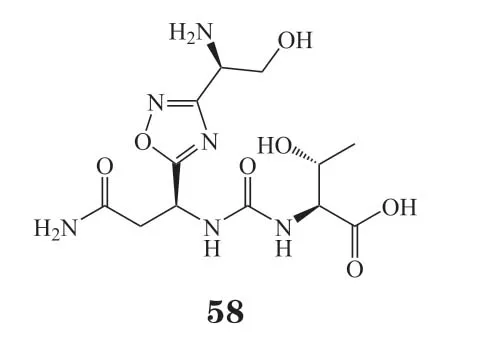
区别于以上的结构类型,Aurigene公司开发了一种新型的噁二唑类PD-1/PD-L1小分子抑制剂CA-170(58),Ⅰ期临床试验结果显示其可显著改善肿瘤患者的病情[97],现已进入Ⅱ期临床试验(NCT01288911)。
4 结语与展望
靶向调控TAMs的抗肿瘤药物在肿瘤免疫治疗领域取得了重大进展,已经确证了多个信号通路在TAMs的趋化、增殖、极化和吞噬过程中发挥关键调节作用。针对这些信号通路的小分子药物的研发也取得了重大突破,尤其是促进TAMs重编程,逆转免疫抑制微环境的CSF-1R抑制剂,已经有多种小分子抑制剂进入临床研究用于多种恶性肿瘤的治疗,其中PLX3397已经成功上市用于腱鞘巨细胞瘤的治疗。在增强TAMs吞噬作用的免疫检查点小分抑制剂的研究方面,由于靶蛋白没有明确的结合口袋,直接结合小分子抑制剂的开发难度极大,目前还处于起步阶段,进入临床研究的小分子抑制剂极其有限,研发状态亟待破冰。
靶向调控TAMs的小分子抑制剂除了用于实体瘤的单一治疗,还可以与T细胞免疫检查点抑制剂联合使用,通过逆转免疫抑制微环境,增加肿瘤组织中T细胞的浸润和杀伤力,提高免疫治疗的疗效和适用范围,有效克服了肿瘤免疫逃逸的难题。目前CSF-1R抑制剂与PD-1或PD-L1单抗联合使用治疗实体瘤的研究正在开展多项临床试验。调控TAMs的不同靶点的联合治疗主要是一些抗体药物,小分子抑制剂的联合治疗鲜有报道,具有深入研究的空间。
综上所述,基于TAMs开发促进肿瘤免疫微环境重塑的全新药物,对提高免疫治疗的临床疗效具有重要的意义,为肿瘤的治疗开辟新的思路和途径。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
第一财经(2019年8期)2019-08-26
海南医学(2016年8期)2016-06-08
池州学院学报(2015年3期)2016-01-05
中国社区医师(2015年14期)2015-12-24
哈尔滨医药(2015年2期)2015-12-01
天津科技大学学报(2015年2期)2015-08-09
学习月刊(2015年14期)2015-07-09
物理化学学报(2015年5期)2015-02-28
