
Ginsenoside Rk2,a dehydroprotopanaxadiol saponin,alleviates alcoholic liver disease via regulating NLRP3 and NLRP6 inflammasome signaling pathways in mice
2023-10-24 02:40JianZouRujieYangRuibingFengJiayueLiuJianBoWan
Jian Zou,Rujie Yang,Ruibing Feng,Jiayue Liu,Jian-Bo Wan
State Key Laboratory of Quality Research in Chinese Medicine,Institute of Chinese Medical Sciences,University of Macau,Macao SAR,China
Keywords:Alcoholic liver disease Ginsenoside Rk2 NLRP3 inflammasome NLRP6 inflammasome Intestinal barrier dysfunction
ABSTRACT Heavy alcohol consumption results in alcoholic liver disease (ALD)with inadequate therapeutic options.Here,we first report the potential beneficial effects of ginsenoside Rk2 (Rk2),a rare dehydroprotopanaxadiol saponin isolated from streamed ginseng,against alcoholic liver injury in mice.Chronic-plus-single-binge ethanol feeding caused severe liver injury,as manifested by significantly elevated serum aminotransferase levels,hepatic histological changes,increased lipid accumulation,oxidative stress,and inflammation in the liver.These deleterious effects were alleviated by the treatment with Rk2(5 and 30 mg/kg).Acting as an nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inhibitor,Rk2 ameliorates alcohol-induced liver inflammation by inhibiting NLRP3 inflammasome signaling in the liver.Meanwhile,the treatment with Rk2 alleviated the alcohol-induced intestinal barrier dysfunction via enhancing NLRP6 inflammasome in the intestine.Our findings indicate that Rk2 is a promising agent for the prevention and treatment of ALD and other NLPR3-driven diseases.
1.Introduction
Alcoholic liver disease (ALD),resulting from heavy alcohol consumption,has a broad spectrum of liver damage ranging from simple fatty liver to more severe forms,such as steatohepatitis,fibrosis,cirrhosis,and even hepatocellular carcinoma[1,2].Harmful alcohol use accounted for about 3.3 million deaths (5.3% of all deaths) and 5.1% of all disability-adjusted life years worldwide in 2016 [3].Despite the widespread global public health burden,satisfactory medication for the management of ALD is limited in the clinic[4,5].Thus,the discovery of a safe and effective agent for the treatment of ALD is urgently needed.
The nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasomemediated sterile inflammation plays a critical role in the pathogenesis of ALD [6-8].Long-term and heavy alcohol exposure disrupts intestinal barrier integrity and enhances bacterial product leakage from the intestinal lumen to the portal vein,leading to increased circulating and hepatic levels of gut-derived lipopolysaccharide (LPS) [9,10].LPS,acting as a most well-characterized pathogen-associated molecular pattern,activates Kupffer cells in the liver through Toll-like receptor 4 (TLR4) signaling,leading to hepatic inflammation in ALD.This process also serves as a priming step of NLRP3 inflammasome activation,resulting in upregulated expression of inflammasome components,including apoptosisassociated speck-like protein containing a CARD (ASC,an adapter molecule),cysteinyl aspartate specific proteinase-1 (caspase-1),and pro-interleukin (IL)-1β [11].Furthermore,damage-associated molecular patterns (DAMPs),released from the damaged hepatocytes induced by alcohol exposure,evoke the assembly of the NLRP3 inflammasome,activating caspase-1,an enzyme that cleaves pro-IL-1β to an active IL-1β[6].This process offers the second signal of activation of NLRP3 inflammasome.Among these identified DAMPs,adenosine triphosphate(ATP)signaling through purinergic P2X receptor 7 (P2X7R) plays a decisive role in ALD [12].Several synthetic small molecules,including 3,4-methylenedioxy-β-nitrostyrene[13],MCC950[14],CY-09[15],INF39[16],and OLT1117[17],have been identified as NLRP3 inhibitors.Recently,natural products,including oridonin [18] and ginsenoside Rh2 [19],have attracted much attention as potential NLRP3 inhibitors in the prevention and treatment of NLRP3-driven diseases,due to their multiple actions and less adverse effects.
Intestinal barrier dysfunction mainly contributes to increased circulating and hepatic gut-derived LPS in ALD [5].However,the exact molecular mechanisms underlying alcohol-induced intestinal barrier dysfunction have not yet been fully elucidated.Accumulating studies have shown that NLRP6 inflammasome,predominantly expressed in enterocytes and colonic goblet cells,plays an essential role in maintaining intestinal homeostasis[20].Moreover,NLRP6 inflammasome preserves intestinal epithelial integrity by modulating the secretions of antimicrobial peptides(adenosine 5’-monophosphates(AMPs))and homeostatic mucin from goblet cells[21-23].AMPs,the key immunological elements,protect the host against pathogen bacteria,virus,and fungi[24],and mucins mainly serve as a barrier overlying the intestinal epithelium[25].However,the impact of NLRP6 inflammasome in ALD has been controversial,based on very few studies[11,26].A recent study indicated that the enhanced NLRP6 activity reduced steatosis,inflammation,and fibrosis in alcoholic hepatitis[26].Still,the other study claimed that NLRP6 plays a disease-aggravating role in a chronic-plus-binge mouse [11].Therefore,the role of NLRP6 inflammasome in ALD remains to be further clarified.
Panax notoginseng(Burk) F.H.Chen (Araliaceae) is a precious medicinal plant used in Chinese medicine and dual food purposes.Its root has been extensively used as an efficient drug for the prevention and treatment of cardiovascular and hematological diseases [27].The roots [28] and leaves [29] ofPanax notoginsengexhibit protective effects against alcohol-induced liver injury.Ginsenosides were considered the main bioactive components contributing to these actions.Several minor ginsenosides were reported to exert inhibitory effects on NLRP3 inflammasome in a variety of animal models,including depression [30],myocardial infarction [31],and D-galactosamine/LPS-induced liver injury [32].Our pilot study indicated that ginsenoside Rk2 (Rk2) showed the most potent anti-inflammatory activity induced by activation of NLRP3 inflammasome without overt cytotoxicity,among the tested minor ginsenosides (Fig.S1).Rk2 is a rare dehydroprotopanaxadiol saponin isolated from steamed ginseng samples [33].Only one study reported the bioactivity of Rk2 with protective effects against ulcerative colitis in vitro [34].Therefore,this study aims to investigate the probable protective effects of Rk2 against alcoholic liver injury in mice and further elucidate its underlying mechanisms targeting hepatic NLRP3 and intestinal NLRP6 inflammasome signaling pathways.
2.Experimental
2.1.Cell experiments
All ginsenosides,including notoginsenosides Fd and Fe,ginsenosides Rk1,Rk2,Rh3 and Rg5,were purchased from Baoji Herbest Bio-Tech Co.,Ltd.(Baoji,China) for the screening test,and their purities were higher than 98% as measured by high performance liquid chromatography-ultraviolet spectrometry.To screen ginsenosides with the inhibitory activity against NLRP3 inflammasome activation,the human leukemia monocytic cells(THP-1)purchased from ATCC (3-5 passages,Manassas,VA,USA) were cultured in Roswell Park Memorial Institute(RPMI)1640 medium,and primed with 100 nM of phorbol 12-myristate 13-acetate (PMA,Sigma-Aldrich,St.Louis,MO,USA) for 24 h to differentiate into macrophages.Cells were treated with different ginsenosides or MCC950(10 μM;Selleckchem,Houston,TX,USA),a selective NLRP3 inhibitor,for 24 h.The cells were stimulated with 10 μM of Nigericin(Thermo Fisher Scientific Inc.,Waltham,MA,USA)for 3 h to activate the NLRP3 inflammasome.Subsequently,the supernatants and cells were collected to measure IL-1β level and protein expression,respectively.For cytotoxicity assay,PMA-primed THP-1 cells were incubated with ginsenosides at different concentrations for 24 h,and cell viability was determined by a Cell Counting Kit-8 Assay(CCK8;Dojindo,Tokyo,Japan).
2.2.Animals and treatments
The 12-week-old C57BL/6 J male mice(weighing 24-26 g)were provided by Animal Research Core,University of Macau (Macao,China).The National Institute on Alcohol Abuse and Alcoholism(NIAAA) animal model of chronic-plus-binge alcohol feeding was used to induce alcoholic liver injury (Fig.S2) as described previously [10,35,36].Briefly,after 5 days of acclimatization to the Lieber-DeCarli liquid control diet(TROPHIC Animal Feed High-Tech Co.,Ltd.,Nantong,China)ad libitum,mice were randomly assigned into four groups (11-13 per group),i.e.,control group (CON),ethanol-fed (ETH) group,and ethanol plus Rk2 treatment(ETH+Rk2;5 and 30 mg/kg)groups.The mice of three ETH groups were fed a Lieber-DeCarli ethanol (5%,V/V) liquid diet for 10 days.The control group received a control diet containing isocaloric maltose dextrin.Mice in Rk2-treated groups were intraperitoneally injected with Rk2 daily at 5 or 30 mg/kg.On the 11thday,all mice were orally administered with a dose of ethanol (5 g/kg) or isocaloric maltose dextrin,and anesthetized with isoflurane 9 h later to collect the samples,including serum,liver,intestine,and fresh feces.All specimens were snap-frozen and stored at -80°C for further analysis.The animal study protocol was endorsed by the Animal Research Ethics Committee,University of Macau (Macao,China) (Approval No.:UMARE-015-2018).
2.3.Biochemical and histological analysis
The serum enzyme activities of aspartate aminotransferase(AST)and alanine aminotransferase(ALT),and serum levels of triglycerides(TG) and total cholesterol (TC) were measured by the commercial quantitative kits (Nanjing Jiancheng Bioengineering Institute,Nanjing,China).Hepatic ATP level was detected by a quantitative kit(Nanjing Jiancheng Bioengineering Institute)according to the manufacturer's instruction,and the value was normalized to total protein.Histological change of the liver was evaluated by hematoxylin and eosin(H&E)staining as described previously[10,37].
2.4.Measurements of lipid accumulation in the liver
For Oil Red O staining,after being fixed with 4% phosphatebuffered paraformaldehyde for 20 min,the cryosections of the liver tissue(8 μm)were stained with Oil Red O(Sigma-Aldrich)for 10 min,and then stained with hematoxylin for 1 min [36].For hepatic lipid quantification,about 50 mg of liver sample from the same lobe was homogenized with ethanol (1:9,m/V).After centrifugation,the supernatant was subjected to TG and TC measurements using commercial quantitative kits.
2.5.Oxidative stress parameters and cytokines in the liver
Liver tissue from the same lobe was used to prepare liver homogenates in cold normal saline (1:9,m/V).The hepatic levels of glutathione (GSH) and Malondialdehyde (MDA),and enzymatic activities of superoxide dismutase (SOD),glutathione peroxidase(GSH-Px) and glutathione reductase (GR) were measured by their commercial kits (Nanjing Jiancheng Bioengineering Institute).Hepatic levels of inflammatory cytokines,including IL-1β,IL-6,and tumor-necrosis factor-α (TNFα),were detected using their corresponding ELISA MAX™Standard kits (BioLegend,San Diego,CA,USA).All data were normalized against the concentration of total hepatic protein.
2.6.Quantitative polymerase chain reaction (qPCR)
The liver and colon samples were grinded into powder under liquid nitrogen.Total RNA was isolated using RNAiso Plus (Takara,Kyoto,Japan)and reverse transcribed using a HiScript II Select qRT SuperMix (Vazyme Biotech Co.,Ltd.,Nanjing,China).qPCR was conducted using TIANGEN Talent qPCR PreMix (FP209-02,Beijing,China)on an ABI Prism 7500 fast real-time PCR System(Vernon,CA,USA).The gene-specific primers were synthesized by Tsingke Biotechnology Co.,Ltd.(Beijing,China),and their sequences are listed in Table S1.
2.7.Immunoblot analysis
Total protein of the cells,liver or colon tissues from the mice was extracted with radio immunoprecipitation assay(RIPA)lysis buffer(Beyotime,Nanjing,China).An equivalent amount of protein from each group was separated by 8%-10% sodium dodecylpolyacrylamide gel electrophoresis and then electrophoretically transferred onto a polyvinylidene fluoride membrane (Millipore,Bedford,MA,USA).After being blocked with 5% non-fat dry milk,the membranes were incubated with primary antibodies(Table S2)overnight at 4°C and then incubated with horseradish peroxidaseconjugated secondary antibody(1:1000,Cell Signaling Technology,Inc.,Danvers,MA,USA).
2.8.Immunofluorescence assay
The immunofluorescence analysis of the liver and colon tissues was conducted as previously described [36].In brief,the fixed cryostat sections (8 μm) were blocked in 5% (V/V) goat serum in phosphate buffer saline (PBS) containing 0.2% (V/V) Tween-20 at room temperature for 1 h,then were incubated with corresponding primary antibodies overnight at 4°C,followed by Alexa Fluor®488 goat anti-rabbit IgG(1:200;Abcam,Cambridge,UK)or Alexa Fluor®488 goat anti-rat IgG (1:200;Abcam) for 2 h at 37°C in the dark.The nuclei were stained with 4′,6-diamidino-2-phenylindole.
2.9.Cellular thermal shift assay
To examine the possible molecular interactions between Rk2 and NLRP3 protein,a cellular thermal shift assay was conducted as described previously[38,39].Briefly,PMA-primed THP-1 cells were lysed with RIPA lysis buffer for 30 min,and the supernatants were collected and incubated with dimethyl sulfoxide,Rk2 (50 μM),or MCC950 (10 μM) at room temperature for 30 min.Subsequently,each lysate aliquot (50 μL) was distributed to clean tubes and individually heated at the indicated temperatures.Afterward,the supernatants(soluble fraction)were collected and immunoblotted for NLRP3 protein.
2.10.Small interfering RNA (siRNA) transfection
THP-1 cells were plated into a 6-well plate at 1×106cells/well density.After being primed with 100 nM of PMA for 24 h,the cells were transfected with a specific NLRP3 siRNA or non-binding control siRNA using GP-transfect-Mate (GenePharma,Shanghai,China) according to the manufacturer's instructions.The NLRP3 siRNA sequences were as follows: siRNA1,sense 5′-GGAGAGACCUUUAUGAGAATT-3′and antisense 5′-UUCUCAUAAAGGUCUCUCCTT-3′;siRNA2,sense 5′-GCAAAGGGCCAUGGACUAUTT-3′and antisense 5′-AUAGUCCAUGGCCCUUUGCTT-3′;siRNA3,sense 5′-CCUCGGUACUCAGCACUAATT -3′and antisense 5′-UUAGUGCUGAGUACCGAGGTT-3′.After transfection for 48 h,the cells were treated with 50 μM of Rk2 for 24 h and then stimulated with 10 μM of nigericin for 3 h.Cell lysates and medium supernatants were collected for further analysis.
2.11.Molecular docking
The crystal structure of target protein NLRP3(protein data bank(PDB) No.: 3QF2) was retrieved from the PDB database.And molecular structure of Rk2 was obtained from the PubChem compound database[40].We prepared and transferred the protein and ligand files firstly into PDBQT format.Any water molecules were removed from the protein structure,and hydrogen atoms were added accordingly.The Grid Box's dimensions were determined based on the protein's domain.We set a docking pocket to 30 Å×30 Å×30 Å square with a 0.05 nm distance.Autodock Vina 1.2.2 software was utilized to visualize the docking model.
2.12.Liquid chromatography-mass spectrometery (LC-MS) analysis
The fecal sample(50 mg)was homogenized with 500 μL of cold methanol.After 10 min of ultrasonication and centrifugation,the supernatant was evaporated to dryness under nitrogen gas.The residue was reconstructed with 50 μL of methanol,centrifuged at 15,800 g for 15 min,and subjected to an ACQUITYTMultraperformance liquid chromatography coupled with a Waters SYNAPT G2-Si Q-TOF high-definition mass spectrometer (Waters Corp.,Manchester,UK).The taurine was separated using an ACQUITY BEH HILIC column (100 mm×2.1 mm i.d.,1.7 μm).The mobile phase consisted of acetonitrile containing 0.1% (V/V) formic acid (phase A) and 0.1% (V/V) aqueous formic acid (phase B),which was pumped at a flow rate of 0.3 mL/min under the following gradient program: 100% A (0-2 min),100%-45% A(2-4.5 min),and back to 100% A for 3 min.Sample injection volume and temperature were 3 μL and 8°C,respectively.The highresolution mass spectra data were acquired in positive MSEmode ranging fromm/z50 to 1,000.
2.13.Statistical analysis
Statistical analysis was performed with GraphPad Prism 8.0(GraphPad Software,Inc.,San Diego,CA,USA).All numerical data in this study were expressed as means±standard error of mean.The multiple comparisons involving three or more groups were evaluated by one-way analysis of variance with Tukey's post hoc test.APvalue of less than 0.05 was considered to be statistically significant.
3.Results
3.1.Rk2 attenuates ethanol-induced hepatoxicity
Alcohol feeding and the Rk2 (Fig.1A) treatments did not affect the body weight of the mice,which indicated that the caloric intake of the mice in different groups was equal(Fig.S3A).The liver index(liver-to-body ratio) in the ETH group was markedly increased by 18%compared to the control,indicating that ethanol feeding results in severe hepatic lipid accumulation.There is no significant difference in liver index between the ETH and Rk2 treatment groups(Fig.S3B).

Fig.1. Ginsenoside Rk2(Rk2)attenuates ethanol-induced hepatoxicity.(A)Chemical structure of Rk2.(B)Serum enzyme activities of aspartate aminotransferase(AST)and alanine aminotransferase(ALT).(C)Representative images of hematoxylin and eosin(H&E)staining of the liver tissue.Data are shown as individual values with means±standard error of mean (n=11-13).***P <0.001 vs.control group (CON) and ##P <0.01 vs.ethanol-fed (ETH) alone group.ns: not statistically significant.
A chronic-plus-binge ethanol feeding results in severe hepatoxicity,as manifested by significantly elevated serum levels of AST (1.73-fold) and ALT (4.76-fold) of the alcohol-fed group over the control group (Fig.1B).These increases were reduced by the treatment with Rk2,either low-dose (5 mg/kg) or high-dose(30 mg/kg),but no significant difference was observed in AST levels between ETH and low-dose Rk2 groups.The histological analysis of the liver tissue showed that alcohol challenge caused the macro lipid vacuoles in the cytosolic compartment,the destroyed hepatocellular structure,and neutrophil infiltration in hepatocytes.These histological changes were apparently alleviated by the treatment with Rk2 at both doses (Fig.1C).
3.2.Rk2 ameliorates ethanol-induced hepatic steatosis
Hepatic steatosis,featured by fat accumulation in the liver,is the main hallmark of the early stage of ALD [41].To examine the potential protective effects of Rk2 on alcohol-induced steatosis in the liver,Oil Red O staining and lipid quantifications were used to examine hepatic fat accumulation.As illustrated by Oil Red O staining (Fig.2A),mice in the ETH group displayed severe hepatic accumulation of lipid droplets (red),and much fewer and smaller microvesicular steatosis was observed in Rk2 treatment groups,either low-or high-dose.Consistently,alcohol exposure significantly elevated hepatic TG levels by 113% (P<0.001) in mice,which was remarkably reversed by the treatment of Rk2 at 5 and 30 mg/kg (Fig.2B).The serum TG level in Rk2-treated mice(30 mg/kg) was significantly lower than in the ETH alone group(Fig.2B).No significant difference was observed in either serum or hepatic TC levels across the groups (Fig.2C),which was consistent with the previous studies [42].

Fig.2. Ginsenoside Rk2 (Rk2) ameliorates ethanol-induced hepatic steatosis.(A) Representative images of Oil Red O staining.(B) Hepatic and serum triglycerides (TG) levels.(C)Hepatic and serum total cholesterol (TC) levels.(D) Immunoblot analysis of hepatic proteins related to de novo lipid synthesis,including phosphorylated adenosine 5’-monophosphate(AMP)-activated protein kinase(AMPK)(p-AMPK),AMPK,sterol-regulatory element binding protein 1(SREBP-1),phosphorylated acetyl-CoA carboxylase(p-ACC),ACC,fatty acid synthase (FASN),and stearoyl-CoA desaturase (SCD1),and (E) their densitometric analysis (n=3). P-values are displayed above the histogram.Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control,and experiments were repeated thrice.Data shown are individual values with means ± standard error of mean(n=11-13).**P <0.01 and ***P <0.001 vs.control group (CON);##P <0.01 and ###P <0.001 vs.ethanol-fed (ETH) alone group.ns: not statistically significant.
Sterol-regulatory element binding protein 1 (SREBP-1)/AMPactivated protein kinase (AMPK) signaling plays a pivotal role in regulating hepatic lipid homeostasis in ALD [5].To understand possible mechanisms underlying the protective effects of Rk2 on ethanol-induced hepatic steatosis,the protein expression of the genes related to de novo fatty acid synthesis,including SREBP-1,AMPK,acetyl-CoA carboxylase (ACC),fatty acid synthase (FASN),and stearoyl-CoA desaturase 1 (SCD1),in the liver were determined by Western blotting.Alcohol consumption downregulated protein levels of phosphorylated AMPK(p-AMPK)and p-ACC,and upregulated SREBP-1,FASN,and SCD1 in the liver,which was notably reversed in Rk2 treatment groups,especially in the highdose group (Figs.2D and E).These results indicate that Rk2 treatment could reduce hepatic steatosis induced by alcohol challenge,which might be attributed to enhanced SREBP-1/AMPK signaling.
3.3.Rk2 relieves hepatic oxidative stress induced by ethanol exposure via enhancing nuclear factor erythroid 2-related factor(Nrf2)/heme oxygenase-1 (HO-1) signaling pathway
Alcohol-induced oxidative stress contributes to the pathogenesis of alcoholic liver injury.Modulation of the antioxidative pathway may be essential to protect against ALD[43].As shown in Fig.3A,alcohol challenge caused significant oxidative stress damage in the liver,as demonstrated by the increased hepatic MDA,the depleted GSH level,and the decreased activities of enzymatic antioxidants,such as GSH-Px,GR,and SOD.These changes were significantly restored by Rk2 treatment at a high dose but not a low dose.Nrf2 and its regulated protein HO-1 are the primary regulators of intracellular redox homeostasis [44].To investigate the mechanism underlying the protection of Rk2 on alcohol-induced oxidative stress,the hepatic expressions of Nrf2 and HO-1 were examined by immunoblot analysis.The protein expressions of Nrf2 and HO-1 in the liver were notably downregulated upon alcohol exposure over the control group.These proteins were significantly upregulated by the treatment with high-dose Rk2(Figs.3B and C).Our data suggest that Rk2 alleviates hepatic oxidative stress induced by alcohol exposure by modulating Nrf2/HO-1 antioxidant pathway.
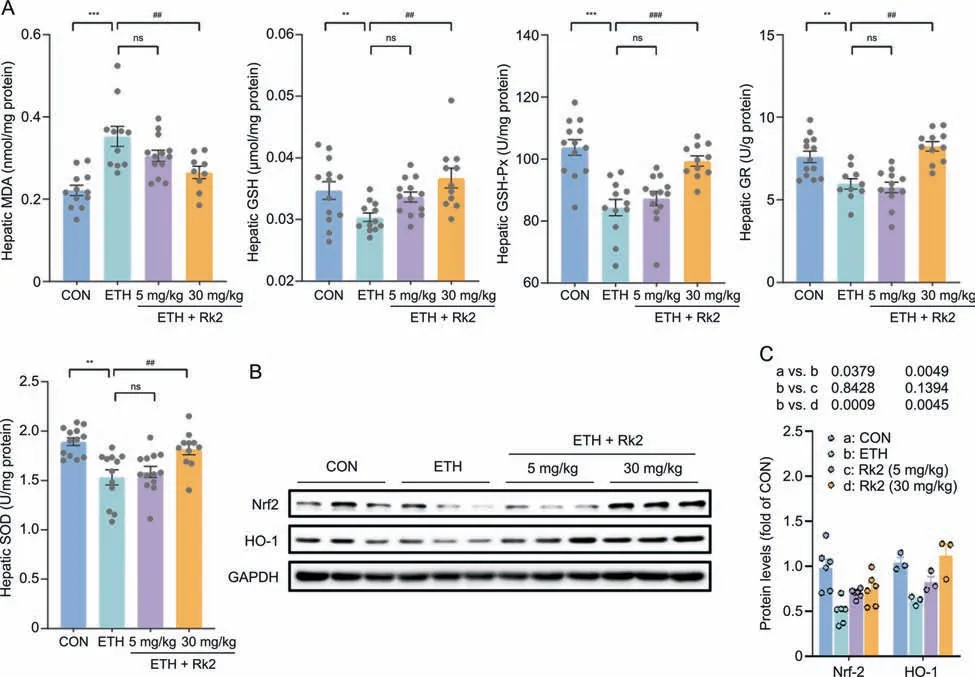
Fig.3. Ginsenoside Rk2 (Rk2) relieves hepatic oxidative stress induced by ethanol exposure via enhancing Nrf-2/HO-1 signaling.(A) Hepatic level of malondialdehyde (MDA),glutathione (GSH),glutathione peroxidase (GSH-Px),glutathione reductase (GR),and superoxide dismutase (SOD).(B) Hepatic protein expression of nuclear factor erythroid 2-related factor (Nrf2) and heme oxygenase-1 (HO-1),and (C) their densitometric analysis (n=3-6). P-values are displayed above the histogram.Glyceraldehyde-3-phosphate dehydrogenase(GAPDH)was used as a control(CON),and experiments were repeated thrice.Data shown are individual values with means±standard error of mean.**P<0.01 and***P <0.001 vs.pair-fed group;##P <0.01 and ###P <0.001 vs.ethanol-fed (ETH) alone group.ns: not statistically significant.
3.4.Rk2 inhibits ethanol-induced hepatic inflammation by inhibiting nuclear factor κB(NF-κB)/NLRP3 inflammasome signaling pathway
To examine the effects of Rk2 on alcohol-induced liver inflammation,the hepatic levels of proinflammatory cytokines,including TNFα,IL-6,and IL-1β,were determined by their commercial kits.As expected,chronic-plus-binge alcohol feeding caused a dramatic increase of TNFα,IL-6,and IL-1β in the liver by 80%,22%,and 26%,respectively,compared to the control group,which was significantly attenuated by the treatment with Rk2 in a dose-dependent manner (Fig.4A).A similar tendency was observed in transcriptional level of IL-1β (Fig.4B).These results were consistent with immunofluorescence assay of F4/80,a cell-surface monocytes/macrophage-specific marker.More number of F4/80-positive cells were observed in the cryosections of liver specimens from the ETH alone group,compared to the control group.Rk2 treatments dosedependently decreased the macrophage infiltration in the liver(Fig.4C).TLR4/NF-κB-mediated activation of Kupffer cells plays a central role in ethanol-induced liver inflammation [45,46].As shown in Figs.4D and S4A,ETH mice showed an apparent increase in protein expressions of TLR4,and its downstream proteins,including MyD88,p-IκBα,and p-P65,compared to the control group.These increased protein expressions were downregulated by Rk2 treatments,especially in the high-dose group.
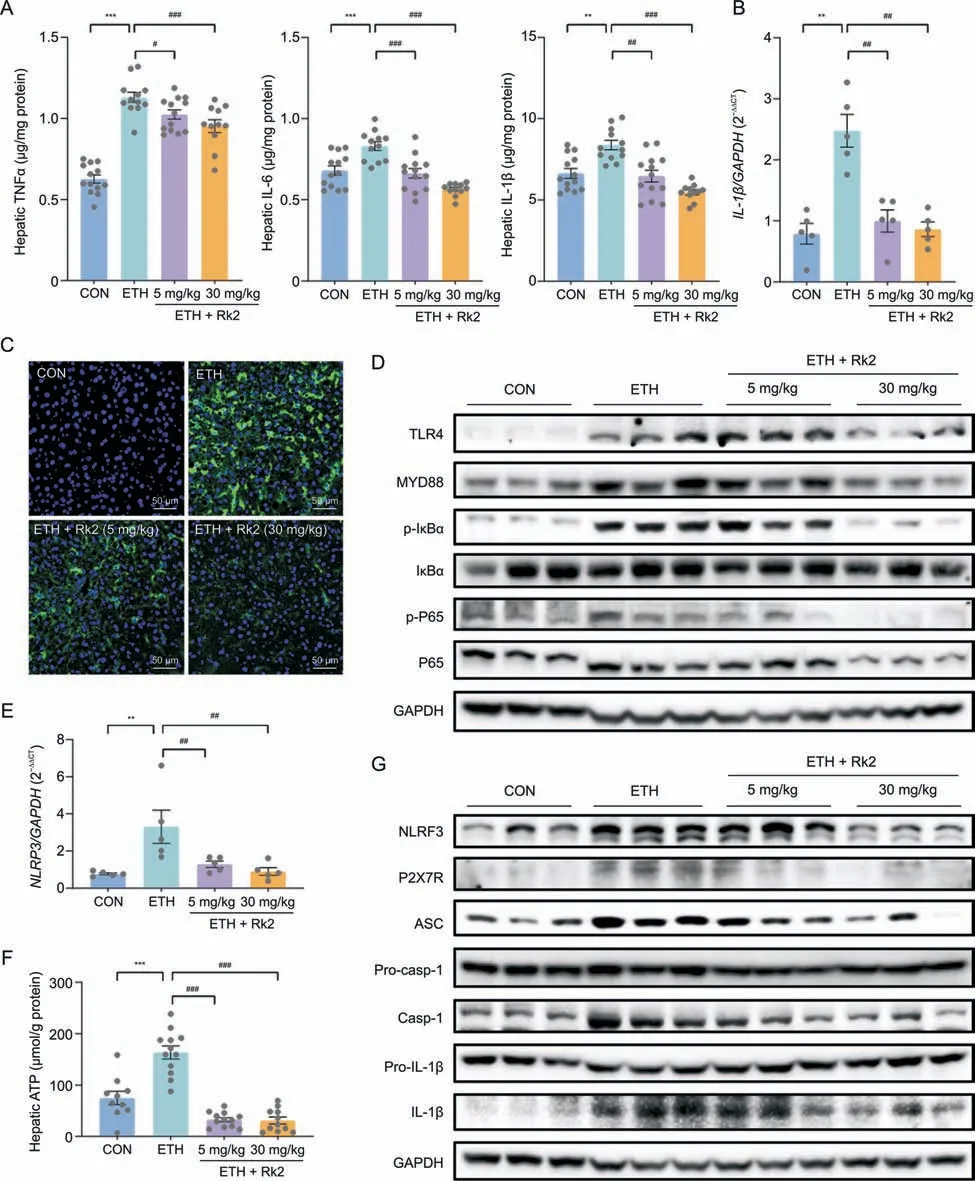
Fig.4. Ginsenoside Rk2(Rk2)inhibits ethanol-induced liver inflammation by inhibiting nuclear factor κB(NF-κB)/nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3(NLRP3)inflammasome signaling pathway.(A)Hepatic level of tumor necrosis factor α(TNFα),interleukin(IL)-6,and IL-1β.(B)Messenger RNA(mRNA)expression of IL-1β.Glyceraldehyde-3-phosphate dehydrogenase(GAPDH)was used as a control.(C)Representative images of immunofluorescence staining of liver sections for F4/80(green).The nucleus was stained with 4',6-diamidino-2-phenylindole (DAPI;blue).(D) Hepatic protein expression of toll-like receptor 4 (TLR4)/myeloid differentiation factor 88(MYD88)/NF-κB signaling pathway.(E) Hepatic mRNA expression of NLRP3. GAPDH was used as a control.(F) Hepatic level of adenosine triphosphate (ATP).(G) Hepatic protein expression of purinergic P2X receptor 7 (P2X7R)/NLRP3 signaling pathway.Experiments were repeated at least three times.**P <0.01 and ***P <0.001 vs.control group (CON);#P<0.05,##P<0.01,and ###P<0.001 vs.ethanol-fed(ETH)alone group.p-IκBα:phosphorylated inhibitor of nuclear factor kappaB α;p-P65:phosphorylated P65;ASC:apoptosisassociated speck-like protein containing a CARD;Pro-casp-1: pro-caspase-1.
The increased hepatic IL-1β level in ALD is primarily attributed to the activation of NLRP3 inflammasome,which plays a crucial role in ethanol-induced hepatic inflammation [47].Our data showed that Rk2 treatment attenuated both protein and messenger RNA(mRNA)expression of hepatic IL-1β induced by alcohol exposure(Figs.4A and B).As shown in Fig.4E,alcohol exposure significantly increased the mRNA expression of NLRP3 in the liver,which was significantly reversed by Rk2 treatments,either low-or high-dose.A similar tendency was observed in the hepatic level of ATP,one of the cytosolic DAMPs that can provoke NLRP3 inflammasome activation (Fig.4F).Furthermore,alcohol exposure obviously upregulated the protein expressions of NLRP3,ASC,caspase-1 and IL-1β in the liver without changing the expressions of pro-caspase-1,and pro-IL-1β,over the control group.These changes were reversed by the treatment with Rk2,especially at 30 mg/kg (Figs.4G and S4B).Collectively,these results suggest that Rk2 inhibits alcohol-induced liver inflammation,which is attributed to inhibiting NLRP3 inflammasome signaling.
3.5.Rk2 acts as a potential NLRP3 inhibitor
As shown in Fig.5A,nigericin significantly induces the secretion of IL-1β in the supernatants of PMA-primed THP-1 cells.10 μM of MCC950,a highly selective inhibitor of NLRP3,dramatically decreased IL-1β level induced by nigericin.The treatment of Rk2 for 24 h at doses ranging from 12.5 μM to 75 μM significantly and dosedependently inhibited nigericin-induced IL-1β secretion,suggesting the potent inhibitory effect of Rk2 on the activation of NLRP3 inflammasome in vitro.Next,we examined the cellular expression of the proteins related to NLRP3 signaling by immunoblot.As shown in Figs.5B and S5A,the protein expressions of NLRP3 and caspase-1 were upregulated in PMA-primed THP-1 cells upon nigericin stimulation,with the decreased pro-caspase-1 expression.These changes were reversed entirely by MCC950 and dosedependently reversed by the treatment of Rk2 (20-75 μM).
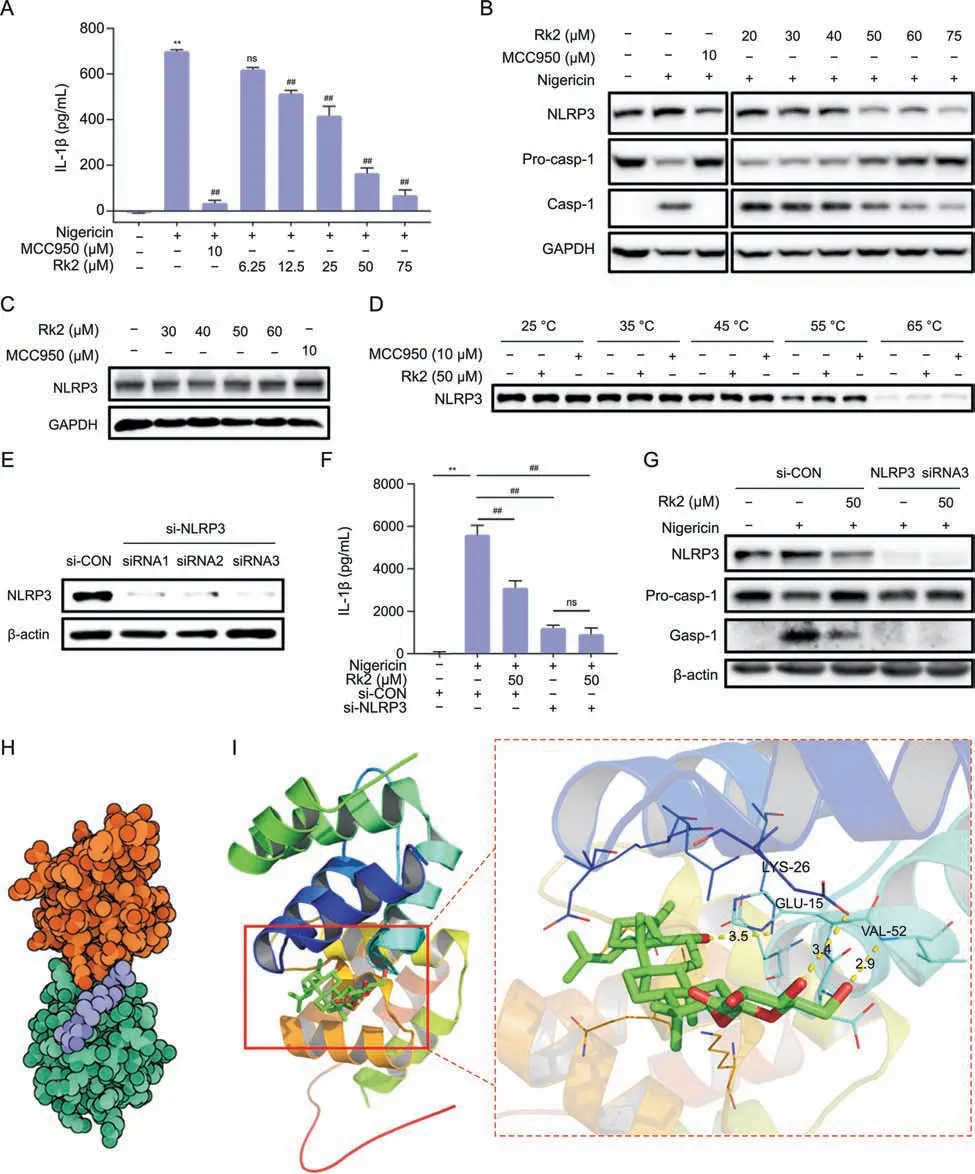
Fig.5. Ginsenoside Rk2(Rk2)acts as a potential nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3(NLRP3)inhibitor.After being treated with MCC950 or Rk2 at indicated concentrations for another 24 h.Phorbol 12-myristate 13-acetate(PMA)-primed THP-1 cells were stimulated with 10 μM of nigericin for 3 h.(A)Interleukin(IL-1β)level in the cell culture medium.(B)Protein expression of NLRP3,pro-caspase-1(Pro-casp-1),and Casp-1.Glyceraldehyde-3-phosphate dehydrogenase(GAPDH)was used as a control.(C) Rk2 alone (30-60 μM) showed no effect on the protein expression of NLRP3 in PMA-primed THP-1 cells.(D) Cellular thermal shift assay.PMA-primed THP-1 cells were transfected with either control small interfering RNA (siRNA) or NLRP3 siRNA for 48 h.(E) The NLRP3 protein levels in the cell lysates were detected using immunoblotting analysis.(F) The cells were treated with Rk2(50 μM) for another 24 h,and then stimulated with nigericin for 3 h,and the IL-1β level in the culture medium was measured.(G) Protein expression of NLRP3,Pro-casp-1,and Casp-1 were measured by immunoblot analysis.(H) The binding mode between Rk2 (purple) and NLRP3 (orange and green).(I)The binding mode of Rk2(green and red)in the binding site of NLRP3(shown in ribbon representation,left),the hydrogen bonding interaction between Rk2(Green and red)and amino acid residues LYS-26,GLU-15,and VAL-52 in the binding pocket of NLRP3(Right).Experiments were repeated at least three times.Data shown are individual values with means ± standard error of mean.**P <0.01 vs.control group (CON);##P <0.01 vs.nigericin-treated group.ns: not statistically significant.
Rk2 alone (30-60 μM) did not affect the protein expression of NLRP3 in PMA-primed THP-1 cells (Figs.5C and S5B).To examine the potential molecular interactions between Rk2 and NLRP3 protein,a cellular thermal shift assay was conducted.As shown in Figs.5D and S5C,the protein expression of NLRP3 in PMA-primed THP-1 cells with or without Rk2 (50 μM)/MCC950 (10 μM) decreased gradually with the increase in heating temperature.Like MCC950,Rk2 can stabilize NLRP3 in the cellular lysates,as evidenced by higher expression of NLRP3 in the Rk2-treated group over the control group,especially at the temperature of 55°C and 65°C.To further validate the interaction between Rk2 and NLRP3,we used siRNA technology to examine the role of NLRP3 in the inhibitory effect of Rk2 on NLRP3 inflammasome activation.As depicted in Figs.5E and S5D,compared with the control siRNA group,PMAprimed THP-1 cells transfected with three different kinds of NLRP3 siRNA all showed almost no expression of NLRP3 in the protein level,suggesting the successful depletion of NLRP3 with siRNA transfection.Then the siRNA3 was chosen for further experiments.As shown in Figs.5F,5G,and S5E,in the siRNA control group,Rk2 at 50 μM significantly inhibited nigericin-induced increase of IL-1β production in the cell supernatants(Fig.5F)and the upregulation of NLRP3 and caspase-1 in PMA-primed THP-1 cells(Figs.5G and S5E).As expected,NLRP3 depletion reversed nigericin induced IL-1β production and caspase-1 activation(Figs.5F,5G,and S5E),and no further inhibitory effect was observed in nigericinstimulated cells treated with Rk2 in the NLRP3 siRNA transfected cells either on IL-1β production or caspase-1 activation(Figs.5F,5G,and S5E).These results indicate that Rk2 might target NLRP3 to inhibit the activation of NLRP3 inflammasome.
To assess the binding energy and interaction mode between Rk2 and NLRP3,we adopted Autodock Vina 1.2.2 software to perform protein-ligand docking.The binding poses and interaction of Rk2 with NLRP3 were obtained with Autodock Vina 1.2.2 software,and binding energy for each interaction was generated.Results showed that Rk2 bound well to NLRP3 protein(Fig.5H),forming hydrogen bonds with amino acid residues GLU-15,LYS-26,and VAL-52(Fig.5I).In addition,the free binding energy of ginsenoside and NLRP3 protein was -6.478 kcal/mol,indicating that Rk2 could spontaneously and stably bind with NLRP3 protein.These results indicate that Rk2 might act as an inhibitor of NLRP3.
3.6.Rk2 ameliorates ethanol-induced intestinal barrier dysfunction
The activation of classic NLRP3 inflammasome results from two sequential steps:inflammasome components and procytokines[7].Alcohol or its metabolites disrupts the intestinal barrier,leading to increased LPS leakage in ALD[48].To further understand how Rk2 inhibits alcohol-induced hepatic inflammation,LPS level in the liver and intestinal barrier integrity were examined.Over the control group,ethanol exposure caused a 3.74-fold increase in hepatic LPS,which were significantly decreased by Rk2 treatments in a dose-dependent manner(Fig.6A).Histological analysis of colon tissue indicated that alcohol feeding caused great loss of the epithelial layer and ulceration of villi,and severe damage to intestinal villi structure and the mucosal lining.These histopathological changes were alleviated by the treatments with Rk2,especially at the high dose of 30 mg/kg(Fig.6B).Furthermore,the decreased expression of zonula occludens-1 (ZO-1),occludin,and claudin 4 were observed in colonic tissue from ETH mice,along with the increased expression of claudin 2,a mediator of the leaky gut barrier.These changes were reversed by the treatment of Rk2(Figs.6C and D).Consistently,qPCR analysis of ZO-1 andclaudin-2(Fig.6E)and immunofluorescence staining of ZO-1(Fig.6F)further confirmed the effect of Rk2 on these tight junction expressions.
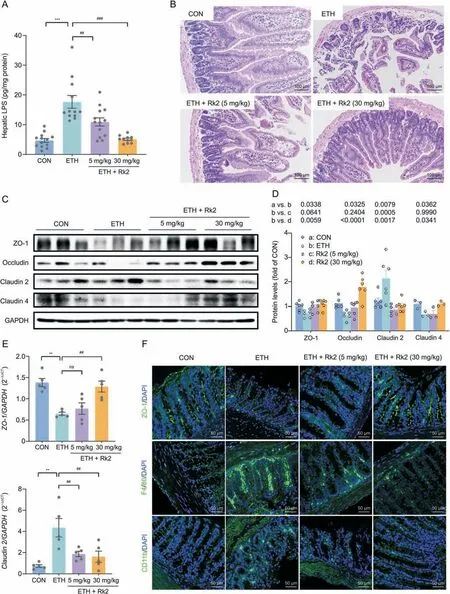
Fig.6. Ginsenoside Rk2 (Rk2) ameliorates ethanol-induced intestinal barrier dysfunction.(A) Hepatic level of lipopolysaccharide (LPS).(B) Representative images of hematoxylin and eosin (H&E) staining of colon tissue.(C) Colonic protein expression of zonula occludens-1 (ZO-1),occludin,claudin-2,and claudin-4,and (D) their densitometric analysis(n=3-6).P-values are displayed above the histogram.Glyceraldehyde-3-phosphate dehydrogenase(GAPDH)was used as a control(CON).(E)Messenger RNA(mRNA)expression of ZO-1 and claudin-2.(F) Representative images of immunofluorescence staining of colonic sections for ZO-1,F4/80,and CD11b.The nucleus was stained with 4',6-diamidino-2-phenylindole(DAPI;blue).Experiments were repeated at least three times.**P <0.01 and ***P<0.001 vs.control group(CON);##P <0.01 and ###P <0.001 vs.ethanol-fed (ETH)alone group.ns: not statistically significant.
Intestinal inflammation contributes to barrier dysfunction in ALD [10].We next examine the inflammatory infiltrate in colon tissue by immunofluorescent staining.As shown in Fig.6F,the intestinal expressions of F4/80 and CD11b,the markers of macrophage and neutrophil,respectively,were significantly increased in the alcohol-fed alone group,which were obviously reversed by the treatments with Rk2,either low-or high-dose.These results indicate that Rk2 treatment restores intestinal barrier dysfunction induced by alcohol exposure.
3.7.The modulation of NLRP6 signaling is implicated in the ameliorative effects of Rk2 on ethanol-induced intestinal barrier dysfunction
Evidence indicates that the NLRP6 inflammasome is critical in maintaining intestinal homeostasis[20].NLRP6 inflammasome activation preserves intestinal barrier integrity through modulating the secretion of AMPs and homeostatic mucin secretion of goblet cells[21,23].To further understand how Rk2 exerts ameliorative effects against ethanol-induced intestinal barrier dysfunction,the possible effects of Rk2 on NLRP6 inflammasome signaling in the colonic tissue were examined.As shown in Fig.7A,the mRNA expression of colonic AMPs,including intestine lectin 1(ITLN1),regenerating islet-derived(REG)-3 lectins,Reg3β,andReg3γ,were markedly downregulated after alcohol exposure,which was significantly reversed by the treatment of high-dose Rk2,but not low-dose.Similar results were observed in mRNA expression ofMUC2that encodes mucin protein MUC2(Fig.7B).Consistently,the histological analysis clearly showed the decreased number of goblet cells in colonic tissue upon alcohol challenge,and these changes were reversed by Rk2 treatments(Fig.6B).Furthermore,ETH mice exhibited a remarkable decrease in colonic protein expressions of NLRP6 and its downstream procaspase-1,caspase-1,and IL-18 as well,a main effector of the NLRP6 inflammasome.These decreased protein expressions were reversed by Rk2 treatments,particularly at 30 mg/kg(Figs.7C and D).The results of colonic NLRP6 expression were further confirmed by qPCR(Fig.7E)and immunofluorescent analysis(Fig.7F).

Fig.7. Nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 6 (NLRP6) signaling is implicated in the ameliorative effects of ginsenoside Rk2(Rk2) on ethanol-induced intestinal barrier dysfunction.(A) Colonic messenger RNA (mRNA) expression of antimicrobial proteins genes intestine lectin 1 (ITLN), regenerating isletderived protein 3β(Reg3β),and Reg3γ,and(B)mucin protein gene(Muc2).Glyceraldehyde-3-phosphate dehydrogenase(GAPDH)was used as a control(CON).(C)Protein expression of NLRP6,pro-caspase-1 (Pro-casp-1) and interleukin (IL)-18 in the colonic tissue,and (D) their densitometric analysis (n=3-6). P-values are displayed above the histogram.(E)Colonic mRNA expression of NLRP6.GAPDH was used as a CON.(F) Representative images of immunofluorescence staining of colon sections for NLRP6 (green),the nucleus was stained with 4',6-diamidino-2-phenylindole (DAPI;blue).(G) Liquid chromatography-mass spectrometery (LC-MS) chromatograms of taurine in the feces of different groups.(H)Semi-quantitative analysis of fecal taurine level(shown as peak area).Experiments were repeated at least three times.*P<0.05 and **P<0.01 vs.CON;#P<0.05 and ##P<0.01 vs.ethanol-fed (ETH) alone group.ns: not statistically significant.
Taurine,a gut microbiota-derived metabolite,is a positive modulator that enhances intestinal NLRP6 inflammasome [23].To further understand how Rk2 activates the NLRP6 signaling,the fecal content of taurine was determined by LC-MS.As shown in Figs.7G and H,ethanol feeding significantly decreased the fecal level of taurine,which was restored by Rk2 treatments in a dose-dependent manner.Although there was no significant difference between the ETH and low-dose Rk2 groups,comparable taurine levels in the feces were observed between the high-dose Rk2 and the control groups.
4.Discussion
Despite ALD having posed a heavy economic burden and severe public health problem,few satisfactory interventions are available for the treatment of ALD [4].In the past decades,natural products have attracted more attention in the management of ALD because of multi-target action and low adverse effects [5].Thus,the discovery of a safe and effective agent from natural products for the treatment of ALD is urgently needed.
Our pilot study indicated that Rk2,a dehydroprotopanaxadiol saponin,showed the most potent inhibitory activity on the cellular inflammation induced by NLRP3 inflammasome activation in PMAprimed THP-1 cells among the minor ginsenosides tested.In this study,the potential effects of Rk2 against alcohol-induced liver injury were revealed in a mouse model of chronic-plus-single-binge ethanol feeding.This NIAAA model simulates the drinking pattern and behaviors of many alcoholics.Furthermore,chronic ethanol feeding for 10 days plus a single ethanol binge can achieve a higher circulating ethanol level and synergistically induce severer hepatic inflammation and liver injury in mice,compared to chronic or acute alcohol consumption alone [49].In our study,the NIAAA model caused severe liver damage in mice,as evidenced by the significant increases in serum levels of aminotransferases,hepatic steatosis,oxidative stress parameters,and hepatic inflammation.These changes were remarkably ameliorated by the intraperitoneal administration of Rk2,especially at 30 mg/kg.Our data,for the first time,indicated that Rk2 ameliorated alcoholic liver injury in mice,which is associated with reduced hepatic inflammation via inhibiting NLRP3 inflammasome in the liver,and the restored intestinal barrier function via enhancing NLRP6 inflammasome in the intestine.
The exacerbating role of NLRP3 inflammasome activation in ALD has been well documented,except for one study reporting the protective effect of NLRP3 inflammasome during alcohol-induced liver injury [9].Chronic alcohol feeding elevated hepatic and serum IL-1β levels and transcriptional expression of NLRP3 inflammasome components,including NLRP3,ASC,and procaspase-1,in mice [50,51].The genetic deficiency of the NLRP3 component,including caspase-1 or ASC,decreased IL-1β levels and ameliorated alcohol-induced liver damage,steatosis,and fibrosis[51].Additionally,the progression of ALD was blunted by the treatment of anakinra,an IL-1R antagonist,or in the IL-1R knockout mice[51].A two-step priming and activation process is essential for NLRP3 inflammasome activation in ALD.In the priming step,alcohol-induced gut-derived LPS activates TLR4/NF-κB signaling,leading to upregulated expression of inflammasome components and pro-IL-1β[7].The second step is assembling and activating the NLRP3 inflammasome,mainly triggered by danger signals released from the damaged hepatocyte,such as ATP and ROS.Extracellular ATP-mediated inflammasome activation through P2X7R has been well-documented to play a crucial role in the pathogenesis of ALD[12,52].The blockage of the P2X7R/NLRP3 inflammasome pathway ameliorates alcoholic hepatosteatosis [53].In this study,alcohol exposure notably upregulated the protein expression of NLRP3 components in the liver,including NLRP3,ASC,caspase-1,and its upstream P2X7R,an ATP receptor.These changes induced by alcohol exposure were reversed upon the treatment of Rk2,especially at 30 mg/kg.The lower levels of hepatic ATP and oxidative stress were observed in the Rk2-treated group compared to the ETH mice.Further study indicated that Rk2 could enhance the thermal stability of NLRP3 in the cellular lysates,especially at the high temperature of 55°C and 65°C.Meanwhile,NLRP3 depletion through siRNA transfection prevented against nigericin-induced caspase-1 activation,and the treatment of Rk2 no longer exerts any effect,suggesting that Rk2 might exert inhibitory effect on NLRP3 inflammasome activation through targeting NLRP3.In addition,molecular docking analysis displayed the hydrogen bonding interactions of Rk2 with NLRP3 protein.Collectively,these results suggest that Rk2,acting as an inhibitor of NLRP3 through binding to NLRP3,ameliorates alcohol-induced liver injury.
Alcohol-induced intestinal barrier dysfunction results in the translocation of bacterial LPS from the gut lumen to the liver through the portal vein,which triggers liver inflammation via TLR4/NF-κB signaling in ALD [54].This process also serves as a priming step of NLRP3 inflammasome activation[11].The circulating level of LPS closely correlates with the severity of alcoholic liver injury[55].Thus,the gut-derived LPS and intestinal barrier could be considered as the potential therapeutic targets of ALD.In this study,the treatment with Rk2 at 30 mg/kg significantly improved intestinal barrier dysfunction induced by alcohol exposure,as manifested by decreased hepatic LPS,alleviated colonic histopathology,and upregulated intestinal tight junction proteins in alcohol-fed mice.
The complex intestinal barrier,consisting of an intact epithelium,various AMPs,and a mucus layer,is the first line of defense to protect healthy intestinal surfaces from adhesion and invasion by luminal microorganisms [56].A large body of evidence has indicated that NLRP6 inflammasome plays a critical role in maintaining intestinal homeostasis via regulating the secretion of AMPs and homeostatic mucin in intestine-related diseases [20,57].However,the impact of NLRP6 inflammasome in ALD is still obscure or controversial [11,26].A study has shown that NLRP6 activation exerts protective effects against steatosis,inflammation,and fibrosis in ALD [26].But a recent finding indicated a diseaseexacerbated role of NLRP6 inflammasome in a chronic-plus-binge mouse [11].Thus,the role of intestinal NLRP6 inflammasome in ALD remains to be further elucidated.In this study,our results indicated that alcohol-fed mice exhibited a remarkable decrease in transcriptional or protein expressions of colonic AMPs,mucin,NLRP6,and its downstream proteins,which is consistent with previous studies [58,59].These changes were restored by Rk2 treatment at the dose of 30 mg/kg,suggesting that the activation of NLRP6 signaling might contribute to the protective effects of Rk2 on intestinal barrier dysfunction induced by ethanol exposure.Taurine,a gut microbiota-derived metabolite,is a positive modulator that enhances intestinal NLRP6 inflammasome-induced IL-18 secretion.No IL-18 induction was observed in taurine-treated ASC-/-or NLRP6-/-mice [23].Furthermore,the increased taurine and decreased histamine by Roux-en-Y gastric bypass surgery significantly improved gut permeability and reduced inflammation in the intestine through reactivating NLRP6 inflammasome [60].In this study,alcohol exposure significantly decreased the taurine level in the fecal sample over the pair-fed mice,which was reversed by Rk2 treatment.The effects of Rk2 on fecal taurine level might be attributed to upregulating taurine-producing gut microbiota,which remains to be further investigated.Taken together,the modulation of the NLRP6 signaling pathway might contribute to the protective effect of Rk2 on ethanol-induced intestinal barrier disruption,partially through regulating intestinal metabolites,such as taurine.
Our current study is the first to reveal that the treatment with Rk2,a rare dehydroprotopanaxadiol saponin isolated from streamed ginseng,effectively ameliorates alcoholic liver injury in mice.This beneficial efficacy is associated with reduced hepatic inflammation via inhibiting NLRP3 inflammasome in the liver and the alleviated intestinal barrier function via enhancing NLRP6 inflammasome in the intestine,as illustrated in Fig.8.Our findings indicate that Rk2,acting as an inhibitor of NLPR3,has a potential to be developed as an efficient agent for the prevention and treatment of ALD and other NLPR3-driven diseases.

Fig.8. Ginsenoside Rk2(Rk2)efficiently ameliorates alcoholic liver disease (ALD)in mice by enhancing intestinal nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 6(NLRP6)and inhibiting hepatic NLRP3 in mice.LPS:lipopolysaccharide;AMP:adenosine 5’-monophosphate;TLR4:toll-like receptor 4;MYD88:myeloid differentiation factor 88;IκBα:phosphorylated inhibitor of nuclear factor kappaB(NF-κB)α;ATP:adenosine triphosphate;P2X7R:purinergic P2X receptor 7;IL:interleukin;AMPK:AMP-activated protein kinase;SREBP-1c: sterol-regulatory element binding protein 1c;ACC: acetyl-CoA carboxylase;FASN: fatty acid synthase;SCD1: stearoyl-CoA desaturase 1.
5.Conclusions
Rk2 regulates fecal taurine content to modulate intestinal NLRP6 expression,which subsequently restores mucus and AMP secretion downregulated by alcohol exposure,thereby improving gut barrier function and reducing LPS leakage to the liver.Meanwhile,as an NLRP3 inhibitor,Rk2 inhibits NLRP3 inflammasome to reduce hepatic inflammation.It also reduces ethanol-induced hepatic steatosis via inhibiting AMPK/SREBP-1 signaling and ameliorates hepatic oxidative stress via enhancing Nrf2/HO-1 anti-oxidant pathway.In conclusion,the treatment of Rk2 efficiently ameliorates ALD in mice by enhancing intestinal NLRP6 and inhibiting hepatic NLRP3 in mice.
CRediT author statement
Jian Zou:Methodology,Validation,Investigation,Data curation,Writing -Original draft preparation;Rujie Yang: Methodology,Investigation,Data curation;Ruibing Feng: Methodology;Jiayue Liu: Resources;Jian-Bo Wan: Conceptualization,Supervision,Writing -Reviewing and Editing;Funding acquisition.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
The work was financially supported by grants from the Research Committee of the University of Macau (Grant No.: MYRG2022-00020-ICMS),and the Science and Technology Development Fund,Macao SAR,China (File No.: 0074/2021/AFJ and 0052/2022/A1).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.05.005.
 Journal of Pharmaceutical Analysis2023年9期
Journal of Pharmaceutical Analysis2023年9期
- Journal of Pharmaceutical Analysis的其它文章
- CLDN18.2-targeted molecular imaging and precision therapy of gastrointestinal tumors
- Applications and safety of gold nanoparticles as therapeutic devices in clinical trials
- Metabolomics: A useful tool for ischemic stroke research
- 1,8-cineole ameliorates colon injury by downregulating macrophage M1 polarization via inhibiting the HSP90-NLRP3-SGT1 complex
- Quantification of soluble epoxide hydrolase inhibitors in experimental and clinical samples using the nanobody-based ELISA
- Gut microbiota-based pharmacokinetic-pharmacodynamic study and molecular mechanism of specnuezhenide in the treatment of colorectal cancer targeting carboxylesterase