
复方克霉唑乳膏中尿素及其缩二脲的质量控制*
2023-10-17 06:24贾微微吴小英卫星红谢子立
中国药业 2023年19期
贾微微,程 杰,吴小英,卫星红,谢子立
(安徽省食品药品检验研究院,安徽合肥 230051)
复方克霉唑乳膏是由尿素、克霉唑与基质组成的消炎抗菌的外用复合制剂。尿素具有抗菌、增加皮肤角质层蛋白质的水合作用,可促进其他药物的经皮吸收[1-2]。国外药典均未收载该品种,其现行标准收载于2020 年版《中国药典(二部)》[3]966,尿素含量测定方法与其收载的尿素乳膏及尿素软膏相同,均采用显色后紫外- 可见分光光度法。尿素分子中伯氨基在酸性条件下与对二甲氨基苯甲醛发生缩合反应,生成黄色的对二甲氨基苯缩脲衍生物。根据埃利希(Ehrlich)反应,对二甲氨基苯甲醛与各种含氮有机化合物作用均会发生显色反应[4-5],但方法专属性差。2020 年版《中国药典(二部)》[3]652、《欧洲药典》[6]中尿素原料药含量测定均采用凯氏定氮法,《美国药典》中采用液相色谱法[7],其他检测方法还有比色法、折光率法、分光光度法、色谱法[8-11]等。复方克霉唑乳膏基质多、辅料多、尿素极性强,导致色谱法分离难度加大,目前未见采用色谱方法测定复方克霉唑乳膏中尿素含量的相关报道。为此,本研究中采用亲水相互作用色谱(HILIC)Amide 色谱柱,结合紫外检测器,建立测定复方克霉唑乳膏中的尿素及其缩二脲含量的高效液相色谱(HPLC)法,同时采用超高效液相色谱-四极杆-静电场轨道阱高分辨质谱(UHPLC-Q-Orbitrap HRMS)法指认尿素的有关物质缩二脲。现报道如下。
1 仪器与试药
1.1 仪器
Q Exactive Focus型四极杆- 高分辨轨道阱液质联用仪(美国Thermo Fisher Scientific 公司);UV - 2700 型紫外- 可见分光光度计,LC - 40 AT 型高效液相色谱仪,均购自日本Shimadzu 公司;XS 105DU 型电子天平(瑞士Mettler Toledo公司,精度为十万分之一);PURA 22 型水浴锅(德国Julabo 公司);FD 240-UL 型电热干燥箱(德国Binder公司)。
1.2 试药
乙腈(色谱级/质谱级,美国Thermo Fisher Scientific公司);甲酸(色谱纯,天津光复精细化工研究所);水为超纯水;尿素对照品(中国食品药品检定研究院,批号为100288-201903,含量为99.8%);缩二脲对照品(上海阿拉丁生化科技股份有限公司,批号为C2011021,含量为99%);复方克霉唑乳膏(A 厂家,共27 批次,其中批号为201003 用于方法学;B 厂家,共27 批次;C 厂家,共3 批次;D 厂家,共2 批次);尿素原料药(A 厂家,批号为 200806;B 厂家,批号为C01120 201007;C 厂家,批号为004120200902);空白辅料由各厂家提供。
2 方法与结果
2.1 色谱与质谱条件
2.1.1 色谱条件
色谱柱:Welch Ultimate HILIC Amide 柱(250 mm ×4.6 mm,5.0µm);流动相:乙腈-0.1%甲酸溶液(95∶5,V/V);流速:0.8 mL/ min;检测波长:195 nm;柱温:35 ℃;进样量:10µL。
2.1.2 质谱条件
离子源:高频火花诱导断裂电离(HESI)源;检测模式:正离子;鞘气压力:206.8 kPa;辅助气体流量:10 mL/ min;喷雾电压:3.80 kV;离子传输管温度:300 ℃;辅助气温度:320 ℃;扫描模式:一级全扫描/数据依赖二级扫描(Full MS/dd-MS2)模式;分辨率:Full MS为70 000,dd - MS2为17 500;扫描范围:55.000 00~825.000 00 质荷比(m/z);隔离窗口:3.0m/z;二级质谱(MS/MS)模式碰撞能量:30/40/50 eV。
2.2 溶液制备
混合对照品溶液:取尿素对照品与缩二脲对照品各适量,精密称定,加乙醇溶解并稀释成每1 mL含尿素0.5 mg、缩二脲2.5µg的溶液,即得。
供试品溶液:取样品适量,精密称定(约相当于尿素50 mg),置100 mL容量瓶中,加乙醇50 mL,水浴加热使尿素溶解,放冷,用乙醇定容,摇匀,冰浴冷却30 min,滤膜滤过,取续滤液,放至室温,即得。
阴性对照品溶液:取A 厂家的空白辅料0.33 g,精密称定,置100 mL 容量瓶中,精密加入克霉唑原料5 mg,加乙醇50 mL,热浴加热15 min,放冷,加乙醇定容,摇匀,冰浴冷却30 min,滤膜滤过,取续滤液,放至室温,即得。
2.3 缩二脲的质谱确认
取2.2 项下供试品溶液适量,按2.1 项下质谱条件测定,确认缩二脲的结构。缩二脲的准分子离子峰为m/z104.045 97[M + H]+,该加氢的准分子离子信号与缩二脲(精确分子量103.038 18)的分子式C2H5N3O2相匹配(质量偏差为0.6×10-6)。由图1可知,二级质谱图中m/z87.019 94 为母离子氨基加氢合物经脱氨气(NH3)后得到,母离子羰基加氢合物经断裂酰胺键后得到m/z61.040 50碎片。

图1 缩二脲的二级质谱分析图Fig.1 Secondary mass spectrometry of biuret
2.4 方法学考察
专属性试验:取2.2 项下混合对照品溶液、供试品溶液及阴性对照品溶液,按2.1 项下色谱条件进样测定。结果显示,溶剂无响应,阴性对照品溶液不干扰尿素及缩二脲的测定;尿素与缩二脲的色谱峰能实现完全分离,两者间的分离度大于8。色谱图见图2。
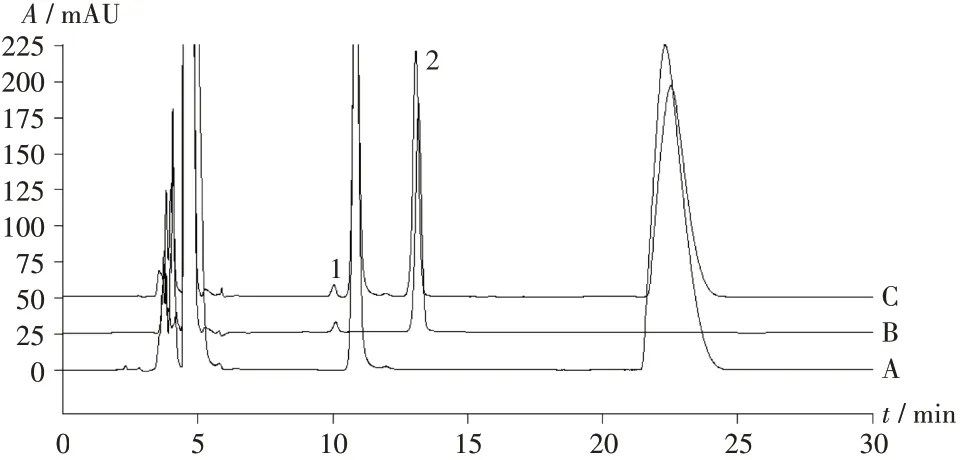
图2 专属性试验高效液相色谱图1.Biuret 2.UreaA.Negative reference solution B.Mixed reference solution C.Test solutionFig.2 HPLC chromatogram of the specificity test
破坏性试验:1)酸破坏。取乳膏内容物,精密称定(约相当于尿素50 mg),置100 mL 容量瓶中,精密加入2.0 mol/L 盐酸1.0 mL,60 ℃放置2 h,放冷,精密加入2.0 mol/L NaOH 溶液1.0 mL,加乙醇50 mL,水浴加热使尿素溶解,放冷,加乙醇定容,摇匀,冰浴冷却30 min,滤膜滤过,取续滤液放至室温。2)碱破坏。取乳膏内容物,精密称定(约相当于尿素50 mg),置100 mL 容量瓶中,精密加入2.0 mol/L NaOH 溶液1.0 mL,60 ℃放置2 h,放冷,精密加入2.0 mol/ L 盐酸1.0 mL,加乙醇50 mL,水浴加热使尿素溶解,放冷,加乙醇定容,摇匀,冰浴冷却30 min,滤膜滤过,取续滤液,放至室温。3)氧化破坏。取乳膏内容物,精密称定(约相当于尿素50 mg),置100 mL 容量瓶中,精密加入30%过氧化氢溶液1.0 mL,60 ℃放置2 h,放冷,加乙醇50 mL,水浴加热使尿素溶解,放冷,加乙醇定容,摇匀,冰浴冷却30 min,滤膜滤过,取续滤液,放至室温。4)光照破坏。直接取未破坏的乳膏供试品溶液,于3 500 lx照射3 d,即得。5)高温破坏。取乳膏内容物2 g,置顶空瓶密封后,105 ℃放置3 h,放冷,精密称定(约相当于尿素50 mg),置100 mL容量瓶中,加乙醇50 mL,水浴加热使尿素溶解,放冷,加乙醇定容,摇匀,冰浴冷却30 min,滤膜滤过,取续滤液,放至室温。分别取上述破坏性试验溶液,按2.1项下色谱条件进样测定,结果尿素及缩二脲在酸、碱、氧化、光破坏条件下均较稳定,在高温破坏条件下缩二脲含量明显增加。色谱图见图3。

图3 破坏性试验高效液相色谱图1.Biuret 2.Ureaa.Destroyed by high temperature b.Destroyed by acid c.Destroyed by alkali d.Destroyed by light e.Destroyed by oxidationFig.3 HPLC chromatogram of the destructive test
线性关系考察:取尿素对照品254.9 mg,精密称定,置50 mL容量瓶中,加乙醇溶解并定容,作为尿素贮备液,并定量稀释成尿素质量浓度分别0.050 9,10.101 8,0.254 4,0.508 8,1.017 6 mg/mL的系列线性溶液;取缩二脲对照品11.57 mg,精密称定,置500 mL容量瓶中,加乙醇溶解并定容,作为缩二脲贮备液,并定量稀释成缩二脲质量浓度分别为0.114 5,0.458 2,1.145 4,2.290 9,4.581 7 µg/ mL 的系列线性溶液。精密量取线性溶液各10µL,按2.1 项下色谱条件进样测定,以质量浓度(C)为横坐标、待测组分峰面积(A)为纵坐标进行线性回归。结果见表1。

表1 线性关系考察结果(n=5)Tab.1 Results of the linear relation test(n=5)
检测限与定量限确定:取2.1 项下对照品溶液,逐级稀释后进样测定,以色谱图中信噪比(S/ N)分别为3∶1和10∶1的质量浓度确定检测限和定量限。结果尿素的检测限和定量限分别为0.25,0.65 µg/ mL;缩二脲的检测限和定量限分别为0.05,0.12µg/mL。
精密度试验:取2.2 项下混合对照品溶液,按2.1项下色谱条件连续进样测定6 次。结果尿素、缩二脲色谱峰峰面积的RSD分别为0.10%和0.46%(n= 6),表明仪器精密度良好。
重复性试验:取样品(批号为201003),按2.2 项下方法制备供试品溶液6 份,按2.1 项下色谱条件分别进样测定。结果尿素和缩二脲的平均含量分别为100.60%和0.59%,RSD分别为0.32%和0.68%(n=6),表明方法重复性良好。
稳定性试验:取2.2 项下供试品溶液,分别于0,2,6,12,18,24 h 时按2.1 项下色谱条件进样测定。结果尿素、缩二脲色谱峰峰面积的RSD分别为0.19% 和0.25%(n=6),表明供试品溶液在24 h稳定性良好。
加样回收试验:取样品(批号为201003)9份,精密称定(每份约相当于尿素25 mg),尿素和缩二脲含量分别为100.60%和0.59%,分别置100 mL 容量瓶中,精密加入线性关系考察项下尿素及缩二脲贮备液各4,5,6 mL,平行3份,加乙醇50 mL,水浴加热使溶解,放冷,加乙醇定容,摇匀,冰浴冷却30 min,滤膜滤过,取续滤液,放至室温,按2.1项下色谱条件进样测定。结果见表2。
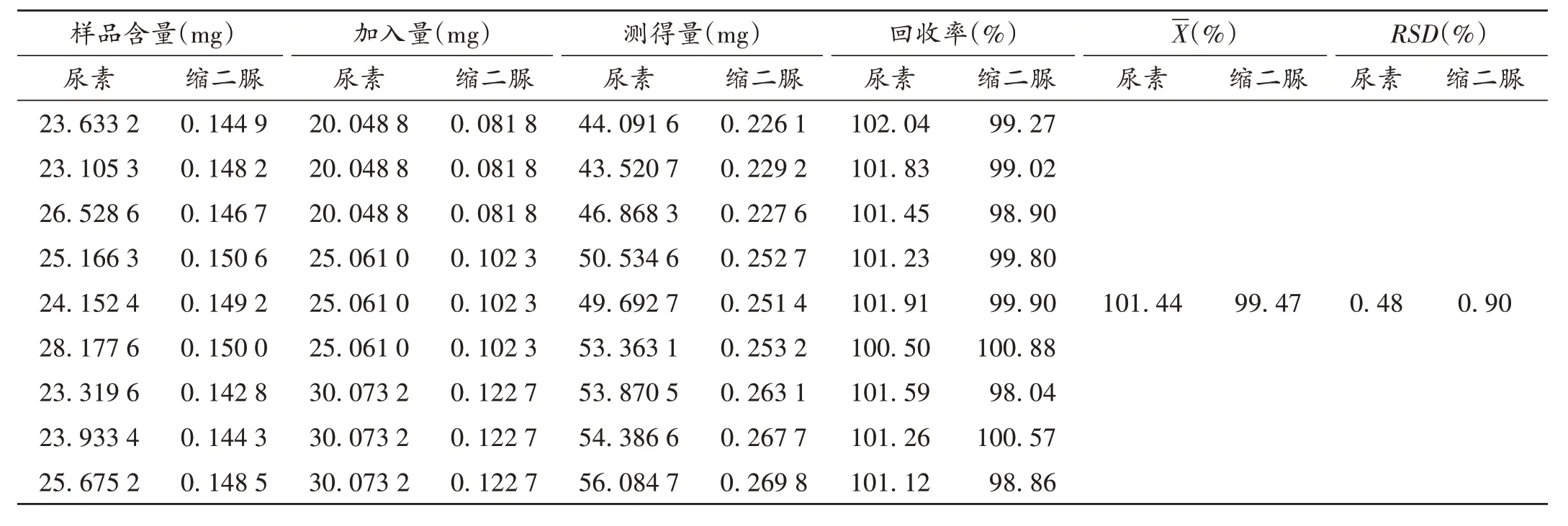
表2 加样回收试验结果(n=9)Tab.2 Results of the recovery test(n=9)
2.5 样品含量测定
尿素含量:按2.1项下拟订方法测定4个厂家59批次复方克霉唑乳膏中尿素的含量,与现行方法(2020 年版《中国药典(二部)》)测得含量进行比较。结果见图4。采用SPSS 19.0 统计学软件分析2 种方法的检验结果,差异有统计学意义(t=6.265,P=0.000 001)。
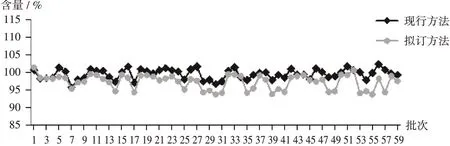
图4 不同方法测得尿素含量比较Fig.4 Comparison of the urea content determined by different methods
缩二脲含量:按2.1 项下拟订方法测定4 个厂家59 批次复方克霉唑乳膏及3 个厂家3 批尿素原料药中缩二脲的含量,结果见表3。
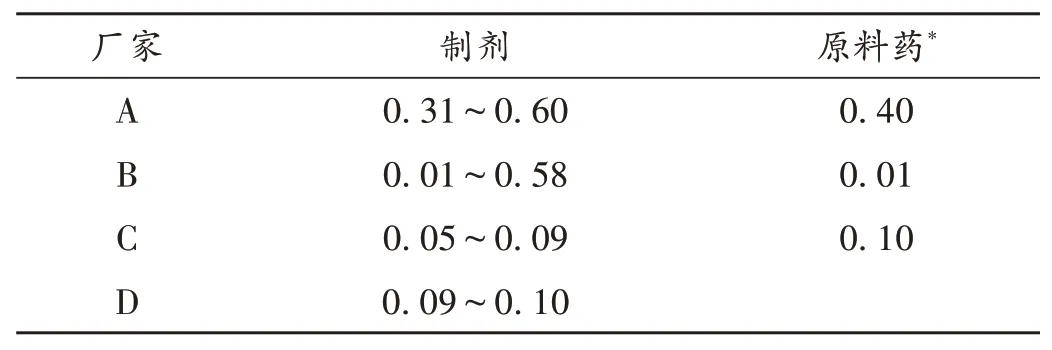
表3 复方克霉唑乳膏与尿素原料药中缩二脲含量测定结果(%)Tab.3 Results of content determination of biuret in Compound Clotrimazole Cream and raw material of urea(%)
3 讨论
3.1 色谱柱、检测波长选择
尿素和缩二脲在反相柱上保留弱,虽有文献报道了使用氨基柱测定尿素[11],但氨基柱柱效下降严重、使用寿命短、耐用性较差。HILIC 柱因其包含了亲水、疏水、分子间作用(如静电、偶极- 偶极、氢键)等在内的复合型原理,在大极性分子的分离、识别、改善峰形等方面优势显著[12-13]。试验考察了HILIC 柱与键合酰胺基作为固定相的HILIC Amide 柱对尿素与缩二脲出峰和分离效果的影响。发现前者虽能满足专属性要求,但基线噪音大,影响了方法的灵敏度,而HILIC Amide 柱满足专属性和灵敏度要求,最终选择HILIC Amide 柱。以流动相作为溶剂,于190~400 nm 波长范围内测定了尿素与缩二脲对照品的紫外光谱图,尿素和缩二脲均末端吸收最大,本研究中参考《美国药典》尿素原料药标准,选择195 nm作为测定波长。
3.2 拟订方法与现行方法测定结果分析
拟订方法较现行方法测定结果偏低的原因是按现行方法检验过程中,克霉唑原料药、克霉唑和尿素的有关物质等各种含氮有机化合物也会与对二甲氨基苯甲醛发生Ehrlich 反应。而2 种方法含量相差4%以上的产品均来自B厂家,可能原因为仅该厂家使用了含氮的辅料依地酸二钠,依地酸二钠中的叔胺基与对二甲氨基苯甲醛发生了显色反应。可见,现行方法不能准确反映药品的实际质量,修订后的尿素含量测定方法更科学、有效和可控。
3.3 缩二脲的形成机制
尿素对热不稳定,100 ℃以上可生成副产物缩二脲[14],这也与高温强制降解试验结果相吻合,同时缩二脲的生成与温度和停留时间成正比[15]。而复方克霉唑乳膏生产中均使用了高温(85~90 ℃)、长反应时间(2~3 h)的油水乳化工艺,故生产工艺中若温度控制不严格也有可能产生工艺杂质缩二脲。
3.4 缩二脲结果分析
B 厂家提供的1 批尿素原料药中缩二脲含量为0.01%,该厂家乳膏中的缩二脲含量在0.01%~0.58%,离散程度大,可能原因为该厂家尿素原料药中的副产物缩二脲含量差异大或乳膏生产过程中温度控制不严格生成了工艺杂质缩二脲。A厂家乳膏中缩二脲含量均较高,与其提供的原料药中缩二脲含量较高相吻合。C 厂家提供的1 批尿素原料药中缩二脲含量为0.10%,而其乳膏中缩二脲含量均低于0.10%。可见,缩二脲含量低的尿素原料药和稳定的生产工艺可使乳膏中的缩二脲控制在较低水平。
3.5 方法评价
本研究中建立的方法操作简便、专属性强、结果准确可靠,可用于复方克霉唑乳膏中尿素及其缩二脲的质量控制,同时也为尿素原料药和以尿素为原料药的制剂的质量控制提供了参考。
猜你喜欢
健康体检与管理(2022年4期)2022-05-13
云南化工(2021年7期)2021-12-21
皮肤病与性病(2021年3期)2021-07-30
健康之家(2021年19期)2021-05-23
中国盐业(2018年20期)2019-01-14
国外医药(抗生素分册)(2016年3期)2016-07-12
中国继续医学教育(2015年4期)2016-01-07
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19
中国药业(2014年21期)2014-05-26
