
1,4-二取代酞嗪衍生物的合成研究进展*
2023-10-12 05:01王蒙蒙张怡天曹明慧王妍姗高艳蓉
化学工程师 2023年9期
王蒙蒙,张怡天,曹明慧,王妍姗,高艳蓉,2
(1.陕西国际商贸学院 医药学院,陕西 咸阳 712046;2.咸阳市分子影像与药物合成重点实验室,陕西 咸阳 712046)
酞嗪是一类重要的含氮芳香杂环化合物,其结构相当于萘环2,3 位的碳原子被氮原子替换而得到的化合物[1]。酞嗪类化合物具有广泛的生物活性,如抗菌抗氧化[2]、抗炎[3]、抗肿瘤[4]、抗高血压等[5]。目前,已经有部分的酞嗪类药物分子上市或处于临床研究阶段,如奥拉帕尼(Olaparib)是一种选择性的PARP1/2 抑制剂,用于治疗基因缺陷相关的晚期卵巢癌(BRCA)[6];布屈嗪(Budralazine)、托屈嗪(Todralazine)及双肼屈嗪(Dihydralazine)等均为治疗高血压的药物[7-9]。作为酞嗪衍生物中具有代表性的一类,1,4-二取代酞嗪衍生物在药物化学领域同样具有重要的用途,并表现出广泛的药理活性,例如,1,4-二取代酞嗪衍生物瓦他拉尼(Vatalanib,PTK787)是一种选择性VEGFR-2 抑制剂,主要用于转移性结直肠癌和前列腺癌的治疗,是1,4-二取代酞嗪衍生物中抗肿瘤药物的优秀代表之一[10](图1)。另外,以Vatalanib 作为先导化合物设计合成得到的1,4-二取代酞嗪衍生物IM-023911 作为VEGFR-2 的有效抑制剂,其IC50达到190nM[11](图1)。

图1 具有代表性的1,4-二取代酞嗪活性分子Fig.1 Representative 1,4-disubstituted phthalazines
正是基于1,4-二取代酞嗪衍生物在药物研发等领域中的重要用途,因此,近年来,开发结构新颖的1,4-二取代酞嗪化合物以及开发新的高效的合成方法已经成为药物化学领域研究的热点课题。通过文献调研,我们根据1,4-二取代酞嗪衍生物的取代基部位结构的差异,将其分为1,4-二氯酞嗪、1-氯-4-氨基酞嗪衍生物、1-烷基-4-氨基酞嗪衍生物、1-芳基-4-氨基酞嗪衍生物及1-芳基-4-烷基或芳基酞嗪衍生物(图2)。然后综述其合成研究进展,期望为1,4-二 取代酞嗪类化合物的合成开发提供参考。

图2 1,4-二取代酞嗪衍生物的结构及分类Fig.2 Structure and classification of 1,4-disubstituted phthalazine derivatives

图3 1,4-二氯酞嗪(3)的合成路线Fig.3 Synthetic route of 1,4-dichlorophthalazine(3)
1 1,4-二取代酞嗪衍生物的合成
1.1 1,4-二氯酞嗪的合成
1,4-二氯酞嗪(3)是一种最简单的1,4-二取代酞嗪,其结构上的两个氯原子可以被其他基团取代。因此,1,4-二氯酞嗪是许多1,4-二取代酞嗪衍生物的关键中间体。王盟盟等[12]报道了1,4-二氯酞嗪的制备方法,该合成路线是以邻苯二甲酸酐(1)为原料,在HOAc 的作用下与水合肼反应生成2,3-二氢酞嗪1,4-二酮(2),再进一步与氯代试剂POCl3反应,生成目标化合物(3),两步反应总收率为85.0%,该合成方法操作简单,适合工业化生产。
1.2 1-氯-4-氨基酞嗪衍生物的合成
1,4-二氯酞嗪(3)作为一种重要的1,4-二取代酞嗪衍生物的关键中间体,可以和不同的亲核试剂发生芳基亲核取代(SNAr)反应制备1-氯-4-氨基酞嗪衍生物。王超杰等[13]报道以邻苯二甲酸酐与水合肼为原料,先发生环合,再经氯代,得到关键中间体1,4-二氯酞嗪(3),然后再与水合肼发生SNAr 反应,实现1,4-二氯酞嗪(3)的单取代,得到1-氯-4-肼酞嗪(4)(图4)。庞园园等[14]报道以1,4-二氯酞嗪(3)为原料、N,N-二甲基甲酰胺(DMF)和水为混合溶剂,在K2CO3碱性条件下,1,4-二氯酞嗪(3)发生单取代得到4-氯-N,N-二甲基酞嗪-1-胺(5),在该反应过程中,DMF 不仅作为溶剂,同时在碱性加热情况下发生分解,得到二甲氨,然后二甲氨作为亲核试剂与1,4-二氯酞嗪(3)发生单SNAr 反应(图4)。

图4 1-氯-4-氨基酞嗪衍生物(4)和(5)的合成路线Fig.4 Synthetic route of 1-chloro-4-amino phthalazine derivatives(4)and(5)
1.3 1-烷基-4-氨基酞嗪衍生物合成
自从1,4-二取代酞嗪衍生物瓦他拉尼(Vatalanib,图1)被发现是一种具有优异选择性的VEGFR-2 抑制剂后,以Vatalanib 作为先导化合物的研究引起了广泛关注。
Eldehna 等[15]报道合成了两个系列具有VEGFR-2 抑制活性的1-烷基-4-氨基酞嗪衍生物(12)和(13),该合成过程同样以邻苯二甲酸酐(1)为原料,首先,与2-(4-甲氧基苯基)乙酸(6)发生缩合反应得到中间体3-(4-甲氧基苯亚甲基)异苯并呋喃-1(3H)-酮(7),随后,中间体(7)与肼反应得到酞嗪酮衍生物(8),然后,在POCl3作用下发生氯代得到关键中间体1-氯-4-(4-甲氧基苯基)酞嗪(9),然后,中间体(9)分别与取代苯胺化合物(10)及脲衍生物(11)发生取代反应得到目标化合物(12)和(13)。该合成过程虽然实现了两个系列1-烷基-4-氨基酞嗪衍生物的合成,但反应第一步缩合过程反应温度高(200℃),具有一定的危险性(图5)。

图5 1-烷基-4-氨基酞嗪衍生物(12)、(13)的合成路线Fig.5 Synthetic route of 1-alkyl-4-amino phthalazine derivatives(12)and(13)
为了开发高效、高选择性的抗肿瘤活性分子,Zhang 等[16,17]设计并合成了一系列新型的1,4-二取代酞嗪衍生物(21)~(24)。随后对所合成的化合物进行体外抗A549、HT-29 和MDA-MB-231 肿瘤细胞活性筛选,发现该系列化合物具有优异的抗肿瘤活性。该合成过程首先以邻苯二甲酸酐(1)为原料,经NaBH4还原得到异苯并呋喃-1(3H)-酮(14),然后,在碱性条件下,与芳香醛(苯甲醛、4-吡啶甲醛)缩合得到中间体(15)、(16),随后,再与水合肼反应得到酞嗪酮中间体(17)、(18),然后,与POCl3发生氯代反应,得到1-氯-4-烷基酞嗪衍生物(19)、(20),再与哌嗪发生亲核取代反应得到1-烷基-4-氨基酞嗪衍生物(21)、(22),最后与氯乙酰基芳胺反应得到1-烷基-4-氨基酞嗪衍生物(23、24)(图6)。该过程经历6 步反应得到Vatalanib 类似物,该反应中异苯并呋喃-1(3H)-酮(14)与芳香醛缩合产物收率较高,具有一定的应用价值。

图6 1-烷基-4-氨基酞嗪衍生物(21)~(24)的合成路线Fig.6 Synthetic route of 1-alkyl-4-amino phthalazine derivatives(21)~(24)
Zhai 等[18,19]合成了3 个系列1-烷基-4-氨基酞 嗪衍生物(31)~(33)(图7),并用于抗肿瘤活性测试。

图7 1-烷基-4-氨基酞嗪衍生物(31)~(33)的合成路线Fig.7 Synthetic route of 1-alkyl-4-amino phthalazine derivatives(31)~(33)
该合成过程以邻苯二甲酸酐(1)为原料,与丙二酸反应得到邻羧基苯乙酮(25),然后在K2CO3条件下与硫酸二甲酯发生酯化反应,得到2-乙酰基苯甲酸甲酯(26),再与苯基三甲基三溴化铵(PTT)发生溴代反应得到2-(2-溴乙酰基)苯甲酸甲酯(27),化合物27 再与取代苯硫酚反应得到硫醚化合物(28),然后再与水合肼反应,得到硫醚取代的酞嗪酮中间体(29),然后发生氯代得到1-氯-4-烷基酞嗪衍生物(30),再与取代苯胺反应得到1-烷基-4-氨基酞嗪衍生物(31),然后再依次经历两次氧化,分别得到亚砜类1-烷基-4-氨基酞嗪衍生物(32)和砜类1-烷基-4-氨基酞嗪衍生物(33)。该反应过程产物收率高,通过2-(2-溴乙酰基)苯甲酸甲酯(27)与取代硫醚反应引入硫原子,随后,通过氧化得到亚砜和砜类化合物,该合成路线对于实现1-烷基-4-氨基酞嗪衍生物种类的多样性具有重要意义。
1.4 1-芳基-4-氨基酞嗪衍生物合成
为了找到更有效和更经济的抗肿瘤药物,Xin等[20]合成了一系列1-苯基-4-氨基酞嗪衍生物(38)、(40)、(41),并评估了其体外抗增殖活性(图8)。

图8 1-芳基-4-氨基酞嗪衍生物(40、41)的合成路线Fig.8 Synthetic route of 1-aryl-4-aminophthalazine derivatives(40 and 41)
该合成是以2-苯甲酰基苯甲酸(34)为原料,首先和肼反应关环,得到酞嗪酮衍生物(35),然后和P2S5反应,得到硫代酞嗪酮衍生物(36),化合物(36)再与氯乙酰胺化合物(37)反应,得到一系列1-芳基-4-硫醚酞嗪衍生物化合物(38);另一条途径是酞嗪酮衍生物(35)经氯代得到1-氯-4-苯基酞嗪(39),随后再与哌嗪反应得到1-苯基-4-(哌嗪-1-基)酞嗪(40),最后再与苄氯衍生物反应得到1-芳基-4-氨基酞嗪衍生物(41),该合成路线得到一系列活性优异的1,4-二取代酞嗪衍生物,但缺点在于中间体(36)合成中会用到毒性较强的P2S5,另外,1-芳基-4-硫醚酞嗪衍生物化合物(38)在合成过程中收率较低,主要是因为副产物较多。
El-Ghaffar 等[21]通过常规方法合成得到1-芳基-4-氯酞嗪中间体(43),该中间体分别与甲醇钠和取代胺反应得到1-芳基-4-甲氧基酞嗪衍生物(44)和1-芳基-4-氨基酞嗪衍生物(45),中间体(45)分别与水杨醛(46)及葡萄糖(47)反应得到两类结构新颖的1-芳基-4-氨基酞嗪衍生物(48)和(49)(图9)。该合成过程路线简洁,产物收率高,底物适用范围广。
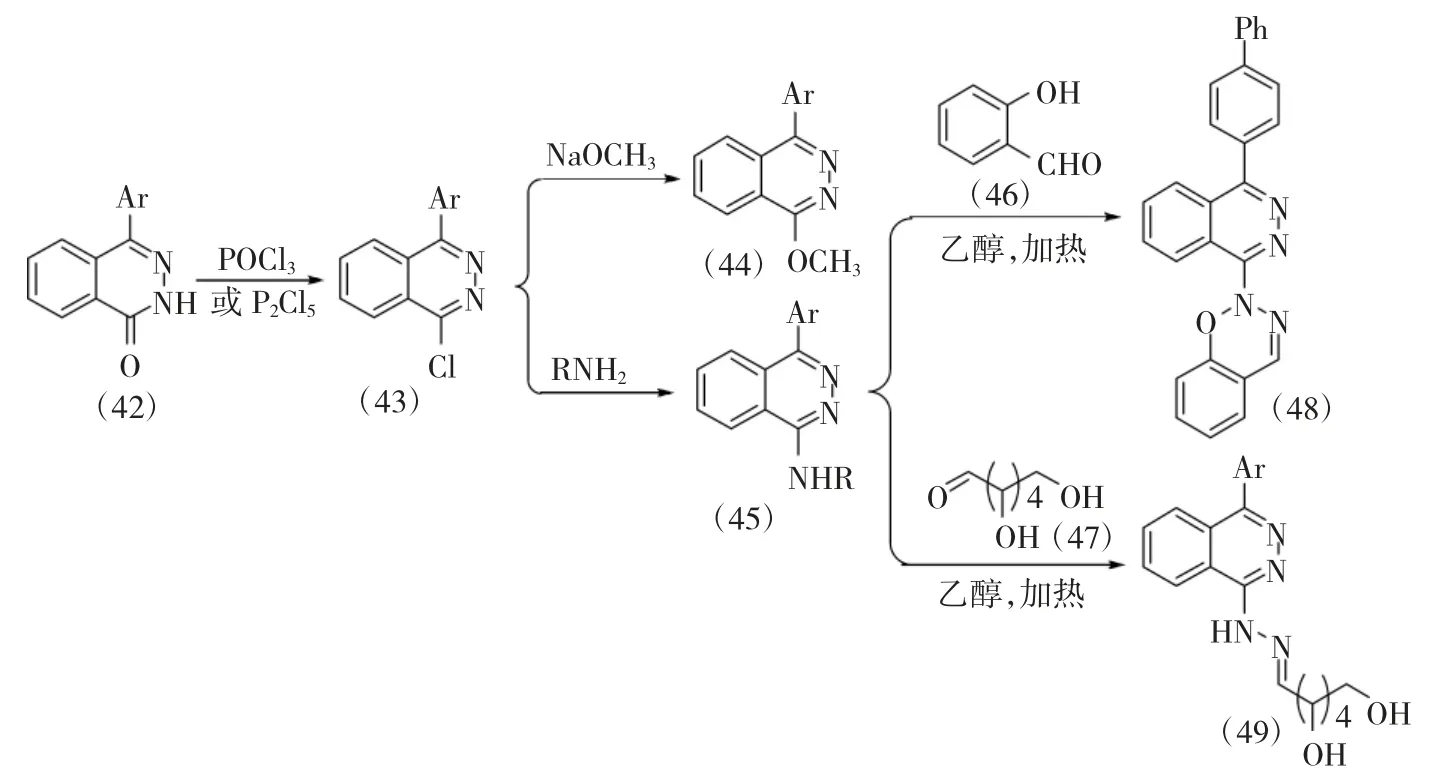
图9 1-芳基-4-氨基酞嗪衍生物(48)和(49)的合成路线Fig.9 Synthetic route of 1-aryl-4-aminophthalazine derivatives(48)and(49)
1.5 1-芳基-4-烷基或芳基酞嗪衍生物合成
Behalo 等[22]报道合成两种1-芳基-4-烷基酞嗪衍生物(53)、(55)和两种1-芳基-4-芳基酞嗪衍生物(54)、(56)(图10)。

图10 1-芳基-4-烷基或芳基酞嗪衍生物(53)~(56)的合成路线Fig.10 Synthetic route of 1-aryl-4-alkyl or aryl phthalazine derivatives(53)~(56)
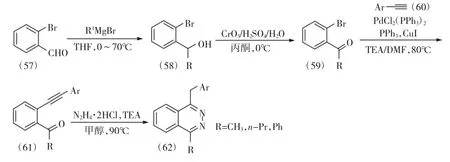
图11 1-芳基-4-烷基或芳基酞嗪衍生物(62)的合成路线Fig.11 Synthetic route of 1-aryl-4-alkyl or aryl phthalazine derivatives(62)
该合成过程首先以邻苯二甲酸酐(1)和二苯醚为原料,在1,1,2,2-四氯乙烷(TCE)溶液中发生Friedel-Crafts 酰基化反应得到2-(4-苯氧基苯甲酰基)苯甲酸(50),随后,依次经历肼关环、氯代得到关键中间体1-氯-4-(4-苯氧基苯基)酞嗪(52)。在强碱性条件下,中间体(52)分别与丙二腈及氰基乙酸乙酯发生芳基亲核取代(SNAr)反应得到两种1-芳基-4-烷基酞嗪衍生物(53)、(55)。(53)和(55)再分别与肼反应,发生关环得到两种含有吡唑取代基的1-芳基-4-芳基酞嗪衍生物(54)、(56)。值得一提的是,该合成路线第一步通过Friedel-Crafts 酰基化反应,较容易得到2-(4-苯氧基苯甲酰基)苯甲酸(50),反应收率也较高。另外,该反应将吡唑杂环及芳环结构引入酞嗪结构1,4 位,可以大大增强产物结构的共轭程度,增强荧光吸收性能。但反应经过芳基亲核取代SNAr 反应制备(53)和(55)的过程中需要加入强碱金属钠,操作具有一定的难度和危险性。
为了构建结构新颖的1-芳基-4-烷基或芳基酞嗪衍生物,廖宗权等[22]报道了一种新的合成方法,首先,以2-溴苯甲醛(57)为原料,分别与不同的格氏试剂(烷基或芳基格氏试剂)发生亲核加成反应得到(58),然后经过琼斯氧化反应将仲醇氧化为酮(59),再与芳基乙炔(60)发生Sonogashira 偶联反应得到2-羰基苯乙炔衍生物(61),最后再与盐酸肼发生插入反应关环得到1-芳基-4-烷基或芳基酞嗪衍生物(62),这一步也是该反应关键步骤,作者通过研究发现,该反应底物适用性广,产物收率高,是合成1,4-二取代酞嗪衍生物的一条有效方法,但该方法的不足之处在于原料2-羰基苯乙炔衍生物(61)的制备过程中需要使用成本较高的钯催化剂,且该偶联反应副产物较多,从而导致收率不高。
2 结论与展望
1,4-二取代酞嗪衍生物在药物化学领域具有重要的用途。因此,对其合成方法开发研究具有重要的价值及意义。本文根据结构差异将1,4-二取代酞嗪衍生物分为5 类,并分别综述其合成研究进展,期望对于开发结构新颖的1,4-二取代酞嗪衍生物提供一定的借鉴作用。目前,虽然已经有大量的1,4-二取代酞嗪衍生物被合成并用于活性研究,但只有较少的几种方法用于合成1,4-二取代酞嗪母核结构,最常见的是通过邻苯二甲酸酐、2-羰基苯甲酸或异苯并呋喃-1-酮衍生物与肼关环得到,但该合成过程通常需要经历多步反应,且产物收率较低;另外,也有通过2-羰基苯乙炔与肼发生插入反应来获得,但该合成方法的不足之处在于原料芳乙炔基化合物制备收率较低,且需要贵金属催化剂等缺点。因此,开发简便高效的合成1,4-二取代酞嗪类衍生物的方法将是该领域的一个重要的研究方向。同时,合成更多1,4-二取代酞嗪类衍生物也将为酞嗪类药物分子的筛选研究提供重要的化合物基础。
猜你喜欢
世界农药(2023年8期)2023-09-04
中国药学药品知识仓库(2022年10期)2022-05-29
云南化工(2021年10期)2021-12-21
汕头大学学报(自然科学版)(2020年4期)2020-12-14
中国资源综合利用(2017年4期)2018-01-22
合成化学(2015年2期)2016-01-17
化工进展(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
无机化学学报(2014年12期)2014-02-28
