
Pd 催化C-X 键跨炔加成的反应机理研究
2023-10-12 04:52范伟菏泽医学专科学校化学教研室山东菏泽274000
化工管理 2023年28期
范伟(菏泽医学专科学校化学教研室,山东 菏泽 274000)
0 引言
对炔键的加成反应是实现对炔键官能团化的一种重要有机合成路径,其具有反应效率高、环境友好、原子经济性好等优点,是近年来过渡金属催化的研究热点[1]。传统的加成反应仅局限于简单的小分子,如:H2、HX、X2、H2O、HCN、NH3、MeOH 等,而且加成反应的形式仅局限于分子间加成,这从一定程度上限制了加成反应的应用范围。随着对合成方法研究的不断深入,科研工作者们研究了Pd、Pt、Fe、Al、Cu、Ni 等多种金属催化的碳-杂键对炔键的加成反应[2],这些加成反应突破了以往的简单小分子对炔键的分子间加成反应的局限,丰富了加成反应的内涵,拓展了加成反应的应用范围。
亚甲基氧吲哚是许多生物活性分子和天然产物中高度相关的基序[3]。尽管它们用于药物化学和天然产物的合成,但以高度立体选择性的方式获得亚甲基氧吲哚的方法是有限的。在此背景下,科研工作者通过Pd(0) 催化炔的碳卤化反应合成了亚甲基氧吲哚,此反应涉及到Pd(II)中C(sp2)-X (I, Br, Cl)的还原消除是关键步骤。虽然芳基卤化物和乙烯基卤化物对Pd(0) 的氧化加成已有广泛研究,并且是Pd 催化中一个相对较好的步骤,但Pd(II) 中C-X 键的还原消除,其优先性并不高。因此,科研工作者们转向了替代方案,例如,从更高氧化态的Pd 中间体(如Pd(III)和Pd(IV))中氧化获得还原消除。通过Pd(II)的还原消除形成C-X 键,在空间要求苛刻的条件下(大体积配体和取代基)和潜在的副反应被抑制(底物控制)的条件下是可行的。不久前,有报道称炔的碳卤化反应中Pd(II)中的C(sp2) -X (I, Br, Cl)得以还原消除(图1),合成方案与醚、烷基兼容。此外,氯氨基甲酰化反应只允许体积较大的三异丙基硅基(TIPS) 炔取代基,若使用体积较小的硅基或体积较大的芳基则无效。这与碳卤化反应(图2)相反,该反应显示出许多具有大体积硅基或芳基炔取代基的芳基卤化物(例如TIPS,1-萘基,9-蒽基)的良好产率。
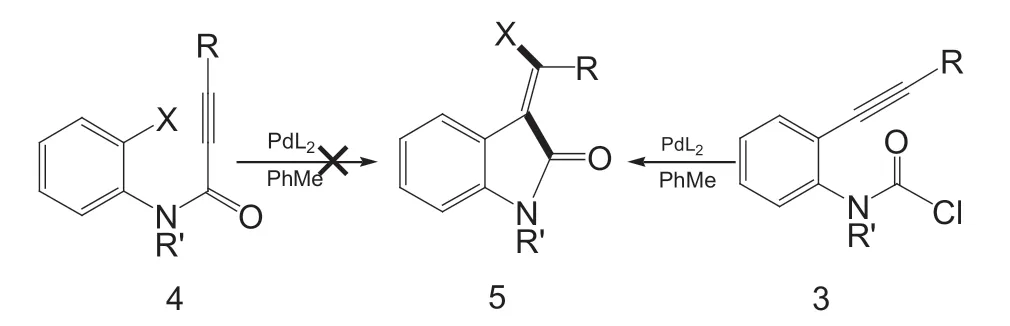
图1 氨基甲酰氯化物的分子内加成

图2 芳基卤化物的分子内加成
为了具体了解配体和底物的选择性、反应性控制因素以及对炔取代基和膦配体的具体要求,利用量化计算结果指导未来的底物和催化反应设计。
1 计算方法
采用Gaussian 09 进行DFT 计算,采用LANL2DZ作为Pd 的ECP,在B3LYP/6-31G(d)理论水平上对气相进行几何优化和频率计算。所有的平稳点都被验证为最小能量或过渡状态。此外,根据固有反应坐标(IRC)确定过渡态(TS)到相应的中间体。在理论的M06L/def2-TZVP 水平上计算能量。
2 结果和讨论
所提出的芳基卤化物和氨基甲酰氯化物分子内加成的机理,根据计算结果,可将芳酰卤化物1 或氨基甲酰氯3 初始氧化加成到单膦Pd(0)中,然后插入炔,直接还原消除(对于芳基卤化物底物1)或快速顺/反异构化和连续还原消除(对于氨甲酰氯底物3),然后分别产生观察到的亚甲基氧吲哚产物Z-2(2a-2d)和E-5(5a 和5b)(图3)。
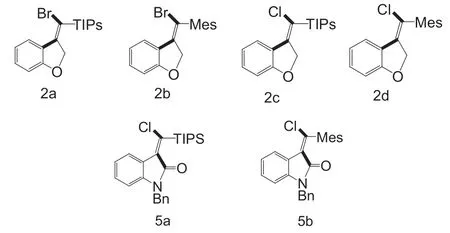
图3 生成物化学式
氨基甲酰氯化物比芳基氯化物反应性强。对C(sp2)-X(Br, Cl) 在炔上的分子内加成的计算表明,芳基卤化物1 或4 的氧化加成是具有最高激活势垒的基本步骤。相反,相应的芳基卤化物在烯烃上的分子内加成是通过还原消除C(sp3)-X(I, Br, Cl)键作为速率决定步骤进行的。在碳酰氯跨炔加成的情况下,发现氧化加成是具有最高激活势垒的TS。
虽然对芳基卤化物的氧化加成已经进行了广泛机理和DFT 计算研究,但对与氨基甲酰氯化物反应的过渡态的性质了解较少。这些物质的高亲电性可能意味着离子、电子转移或形成亲核取代反应。氨基甲酰氯的氧化加成明显优于芳基氯,快速氧化加成步骤据推测是反应的过渡态(图4 所示)。因此,对3a 和4a与Pd/PA-Ph 催化反应的静息状态进行了分析。通过核磁共振观察到氯氨甲酰3a 几乎瞬间转化,同时形成新的含膦物质和游离配体。相比之下,芳酰氯4a 在相同的条件下只产生了微量的产物。此反应条件下,只观察到一种主要的含磷物质,这是催化剂分解的结果。此外,在氨甲酰氯3a 的反应中,在核磁共振谱中形成了一个新物质,该物质随时间的推移而减少,推测是氧化加成中间体。基于这些信息,氯化氨甲酰3a的氧化加成不可能是具有最高激活势垒的初级步骤,而观察到潜在的氧化加成中间体则表明炔插入,即碳化反应是决定转化率的过渡态。因此,氨甲酰氯3a 的氧化加成据推测是通过一个快速的、可能是离子的、亲核的取代反应进行的。根据实验结果,可推测3a 的氧化加成是快速的炔插入,是氨甲酰氯跨炔加成形成的。为了比较氨甲酰氯和芳酰氯在炔间加成反应中的反应活性,计算了完整的反应路径。图5 所示为芳基卤化物和氨基甲酰氯化物在炔上加成的历程以及进一步分析计算出的吉布斯自由能路径和底物3、4 的能量跨度,揭示了配体、卤化物和炔取代基对反应结果的影响。

图4 反应历程图
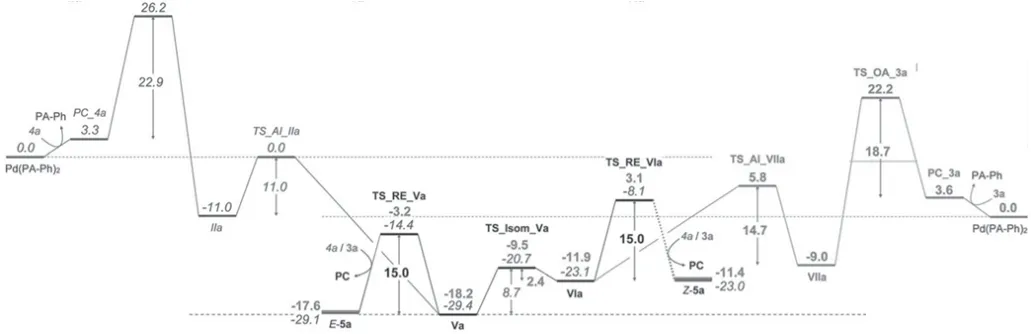
图5 能量曲线图
较高的反应温度会导致催化剂分解,从而阻止催化反应。催化剂的分解是由酰胺部分促进的。计算了Pd(PA-Ph) 氧化插入底物3a 和4a 潜在激活键的过渡态,并比较了相应的激活势垒。实验表明,通过快速的、可能是离子的、亲核的取代,对氨基甲酰氯的氧化加成具有更低的屏障。相比之下,对于第一代芳酰氯底物4a,在类似的反应条件下没有反应,计算表明,炔的C-Si 键的活化与C-Cl 键的氧化添加是竞争的。这一观察结果是由于炔与酰胺的共轭作用增加了C-Si键的反应性。
膦配体的作用:体积庞大、富含电子的磷化氢配体,如PtBu3,非常适合对于在烯烃和炔烃之间加成芳基卤化物,它们在炔之间加成氨基甲酰氯化物时的使用只会形成少量的产物[4]。氧化加成是第一代芳基卤化物,底物4 的速率决定TS,而炔插入是第二代氨基氯底物,3 的速率决定TS。在芳基卤化物底物4 的情况下,体积较大的PtBu3配体促进了产物的还原消除,从而与体积较小的PA-Ph 配体相比,降低了反应的吉布斯自由能。相反,在氨基氯3 的作用下,体积较小的PA-Ph 有利于炔插入,与采用体积较大的PtBu3配体的相应过程相比,其激活降低了约5 千焦每摩尔。从而解释了与体积较大的PtBu3配体相比,PA-Ph 具有优越反应性的原因。
甲硅烷基的效果:在实验中,只有在炔上带有硅基取代基的底物才能在氯氨基甲酰化反应中发生反应,而在类似的反应条件下,带有三甲基取代的炔的底物不能发生环化反应。计算结果表明:当比较底物3a 和3b 的能量途径时,观察到TIPS 部分对顺式/反式异构化和还原消除的影响。TIPS 基团导致Pd(II)中间体的不稳定性,以及顺式、反式异构化过渡态的稳定。这导致异构化难度的降低。
硅基的立体效应:对于TIPS 取代的Pd(II)中间体,反式中间体Va 比对应的顺式中间体VIa 更稳定,而对甲酰基取代的Pd(II)中间体则相反。与相应的甲酰基取代中间体相比,Pd(II)中间体VIa 和Va 都明显不能被TIPS 部分破坏。然而,顺式pd (II)中间体VIa的不稳定程度更为明显,与Va 相比,VIa 中的TIPS 部分与氧吲哚的芳基之间的空间相互作用增加。相反,甲酰基部分可以旋转,其中空间相互作用明显更少,从而使Pd 取代基成为最具空间拥挤性的部分。总的来说,硅基部分在氯氨基甲酰化反应中对几个中间体和步骤产生综合影响,从而产生其独特的反应活性[5]。
选择性差异的起源:在氨甲酰氯化物的反应中,通过快速顺/反异构化,可以达到完全的反式选择性。然而,在相应的芳基卤化物反应中,由于直接还原消除,可以观察到良好的选择性。在甲酰基取代炔的情况下,在较高的反应温度下选择性的转换得到E-2b。此外,底物范围不限于TIPS 取代的炔,也可以是其他集团,例如芳基。
通过计算得到的自由能途径的分析,炔取代基对反应活性有显著的影响。类似于酰胺类底物3 和4 的反应,TIPS 取代基有利于生成顺式Pd (II)中间体,但相应的跨中间体IV 在具有甲酰基时更稳定。
3 结语
通过实验和DFT 计算研究了炔烃碳卤化和氯氨基酰化反应的机理,提出了氧化加成、炔插入、顺/反异构化和还原消除等催化途径。在芳基卤化物跨炔加成的情况下,氧化加成是限制反应活性的;而在相应的氨甲酰氯化物反应中,氧化加成却被证明是加快反应速率的,数据表明,取而代之的是炔插入限制了反应性。研究了卤化物、膦配体和炔取代基对反应活性的影响:体积庞大的PtBu3对于芳基卤化物跨炔加成的分子内反应活性至关重要,降低了还原消除的障碍;体积较小的金刚烷膦配体PA-Ph 特别适合于相应的氨甲酰氯化物加成反应。计算结果表明,与PtBu3相比,PA-Ph 配体的体积较小,因此它限制炔插入的屏障显著降低。炔取代基对反应性有显著影响,这解释了携带TIPS 取代基的底物的特殊反应性。庞大的TIPS 基团会导致Pd(II)中间体V 和VI 的不稳定,以及顺、反异构化TS 的稳定。这总体上导致更小的能量跨度,从而增加催化周转。
这些探究结果将会为今后的反应条件筛选和优化、更加经济高效和高选择性的反应体系的设计提供有价值的参考。
金属催化的C-X 键对炔键的加成反应在近些年取得了重要进展,但是此研究领域仍然有许多挑战[6]。如目前使用的大部分金属催化剂仍是价格较昂贵的Pd、Ru、Rh、Ni、Ir、Pt、Au 等催化剂,而且目前报道的许多类型的碳-杂键对炔键的加成反应仅局限于分子内而不能适用于分子间,此外还有许多类型的碳-杂键对炔键的加成反应尚未实现。相信在不久的将来,在此领域会有更多突破性的进展,使得金属催化的对炔键的加成反应成为对炔键进行官能团化的一种重要策略,并在有机合成中发挥更加重要的作用。
猜你喜欢
环境卫生工程(2021年5期)2021-11-20
发光学报(2021年6期)2021-06-16
分析化学(2019年3期)2019-03-30
上海计量测试(2018年3期)2018-07-09
生物化工(2016年4期)2016-04-08
合成化学(2015年2期)2016-01-17
化工进展(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
山东工业技术(2015年6期)2015-07-27
河北大学学报(自然科学版)(2012年3期)2012-03-25
