
多核超图神经网络单细胞分类算法
2023-10-09 01:47李荣远龙法宁
计算机应用与软件 2023年9期
李荣远 龙法宁
1(广西师范大学计算机科学与工程学院 广西 桂林 541004)
2(玉林师范学院计算机科学与工程学院 广西 玉林 537000)
0 引 言
鉴定细胞类型及亚型成为scRNA-seq重要的应用之一,大量的半监督、无监督聚类方法被开发出来。基于聚类的分类方法假定聚类中所有细胞均属于同一类型,因此可以进行集群标记。但这种假设通常是错误的,集群中除了主要细胞类型外,通常还包含少量占比的多种细胞类型[1]。常用的聚类算法如k-means、hierarchical clustering需要设置相应的类别数,其中类别数的设置对聚类结果影响较大。因此,一种无须先进行聚类就可对每个细胞进行分类的方法可解决该问题。ScRNA-seq测序技术由于本身技术噪声、批次效应等问题,导致下游分析困难,且单细胞表达谱维度较高,至少上万维,普通方法难以分辨细胞类型。超图神经网络能较好地处理大规模多模态数据集[2]。基于此,本文尝试将特征工程结合超图神经网络应用于单细胞测试数据集上来证明方法的有效性。
1 相关工作
继《基因组计划》后,Regev等[3]提出《人类细胞图谱计划》,该计划旨在绘制人类众多细胞类型及状态。计划描述出人类每个细胞,探索先前未知细胞,如研究细胞类型、细胞之间关系和细胞组成成分等,因此多种单细胞RNA测序技术(ScRNA-seq)蜂拥而至。目前主流的微流控技术有10X Genomics和微孔板技术[4]等,其测序通量较高,能对单细胞全长测序,基因检测率较高。ScRNA-seq能测序出单细胞整个转录组的基因表达,方便各种方法进行细胞分类,识别细胞亚型,探索细胞的异质性[5]。
目前单细胞主流研究方向主要有:转录组学、基因组学和轨迹推断[6]等分支。本文主要研究单细胞转录组方向,目前面临一些挑战,单个细胞转录状态的所有表征能全面了解细胞内RNA的相互作用,但scRNA-seq观测值零值较多,给定的基因中没有唯一的分子标识符。例如,缺失率(Dropout)通常被描述为scRNA-seq数据中的零值,但是该项观测通常将两种不同零值类型混为一起。一是归因于噪声,其基因已经表达但未被测序技术检测到的零值;二是归因于生物学上真正的零值。不建议将Dropout作为观察值为零的总称[7-8],这些零值归因于技术限制:可能是生物变异、细胞裂解和人工操作等因素。较多零值使高维数据变得稀疏,稀疏度取决于所用的scRNA-seq平台、测序深度和基因自身表达水平。ScRNA-seq数据的稀疏性可能会阻碍下游分析,难以正确建模或处理,因此需要进一步开发新方法。M3Drop[9]实现两种针对零值的处理方法。第一种方法适合于全转录组测序协议产生的数据;第二种适合数据集符合负二项分布ZINB(零膨胀负二项式)模型NBDrop,它能提取出具有较高零值的数据特征。两种基于Dropout的特征选择方法NBDrop和M3Drop均比基于方差的特征选择方法好,但不适合小样本量或高噪声数据。
特征工程在总体预测性能中起关键作用,包括质量控制(删除细胞、基因)、规范化处理、基因选择(选高变异基因)或降维,对聚类或分类模型起关键作用。文献[10]提出3种基因选择方法及14种聚类算法分析。其中SC3[11]和Seurat[12]表现出最好结果,但SC3运行大数据集耗时较长,效率较低。Seurat在处理scRNA-sq数据集上效果较好,其中特征工程中选出高度变异的基因是其关键。文献[1]提出scPred方法,这是一种新的可推广方法,该方法结合基于降维的特征选择和机器学习的预测方法,对单细胞进行高度准确的分类。但该方法局限于一些特定数据集,对多种单细胞测序平台兼容效果差,分类准确率低。
基于超图的思想在单细胞数据集方面应用较少,SAME[13]使用超图将多种方法进行聚类集成,将聚类标签作为输入,从而构建一种共识机制。SAFE[14]集成四种最先进的聚类方法:SC3、CIDR、Seurat和t-SNE+k-means,该方法运行时间开销较大。
基于标记的单细胞分类必不可少,超图学习[15]在处理大规模数据集有较好效果。超图处理复杂数据更灵活,HGNN根据超边卷积学习数据之间的相关性,有效地进行传统的超图学习。超高维多模态数据符合这种模型处理[16],scRNA-seq数据集正是一种超高维数据集。
综上所述,单细胞多核超图分类整体流程如图1所示。
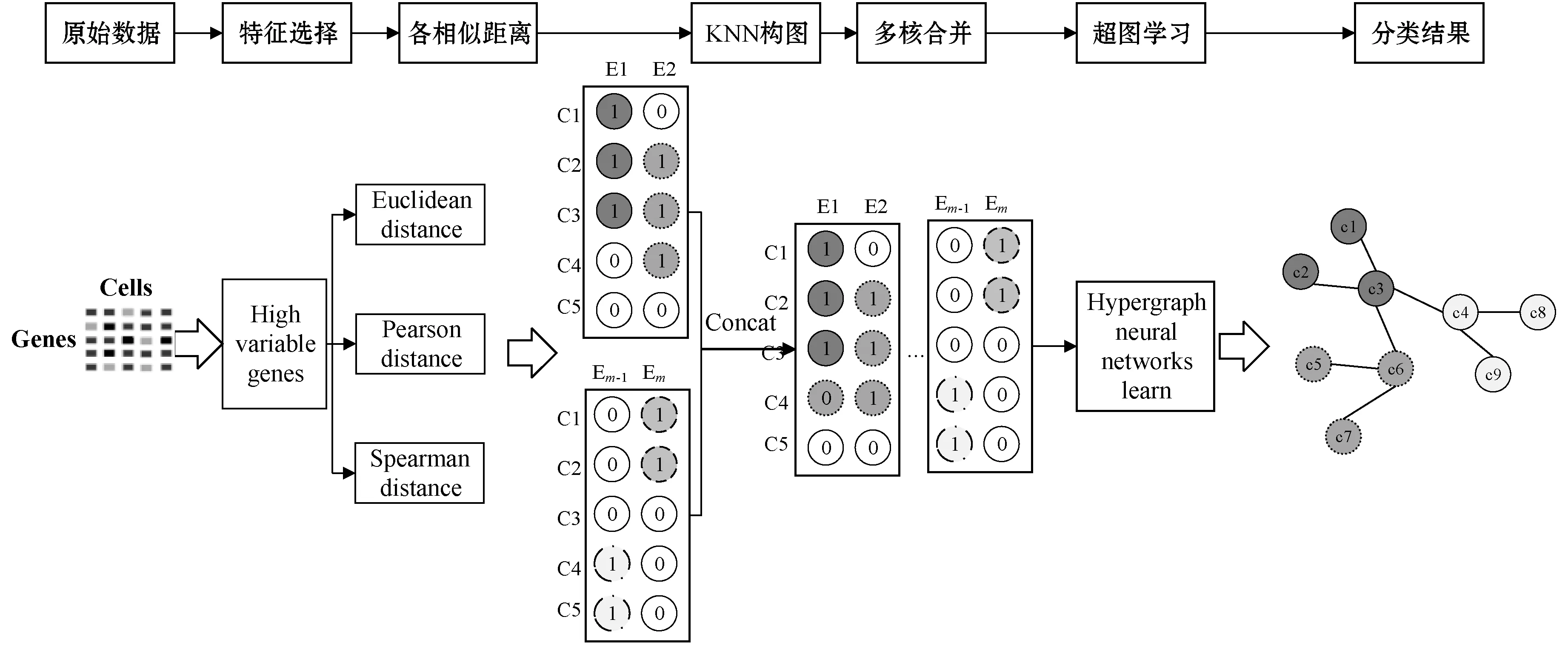
图1 单细胞多核超图分类整体流程
(1) 数据预处理。收集多种平台数据集,将单细胞数据经过预处理后,选出高度变异的基因(High Variable Gene,HVG)。
(2) 多核KNN图构建。计算细胞之间各相似距离,根据距离构建各细胞K近邻图,将多核KNN图合并为超图。
(3) 构建超图神经网络学习HGNN(Hypergraph Neural Network)。通过点-边-点特征表示,构建超图学习分类器。
(4) 实验对比验证。采用多种数据类型进行验证三种方法。主流分类方法有scPred、HGNN和HVG-MHGNN(Multi-kernel Hypergraph Neural Network based on High Variable Gene)。
2 scRNA-seq的HVG-MHGNN模型构建
2.1 scRNA-seq数据集整理
ScRNA-seq测序平台多样,为验证实验方法,收集了多种类型数据。数据集主要来源conquer数据库、10X、GEO、ArrayExpress。Trapnell、TrapnellTCC和Petropoulos来源于conquer。整理多种类型数据集工作量较大,一些作者只提供原始ATGC序列数据集,一般都是上百GB,普通平台难以分析。本次收集作者处理过的基因表达值,最终形成count统计量类型数据。由于一些数据集作者标签隐藏在数据库中,需要查看作者原始论文人工提取标签,以方便验证。如Petropoulos数据集从88个人类植入前胚胎中获得了1 529个单细胞RNA-seq,观察胚胎从3天到第7天发育情况。其他数据集如表1所示。
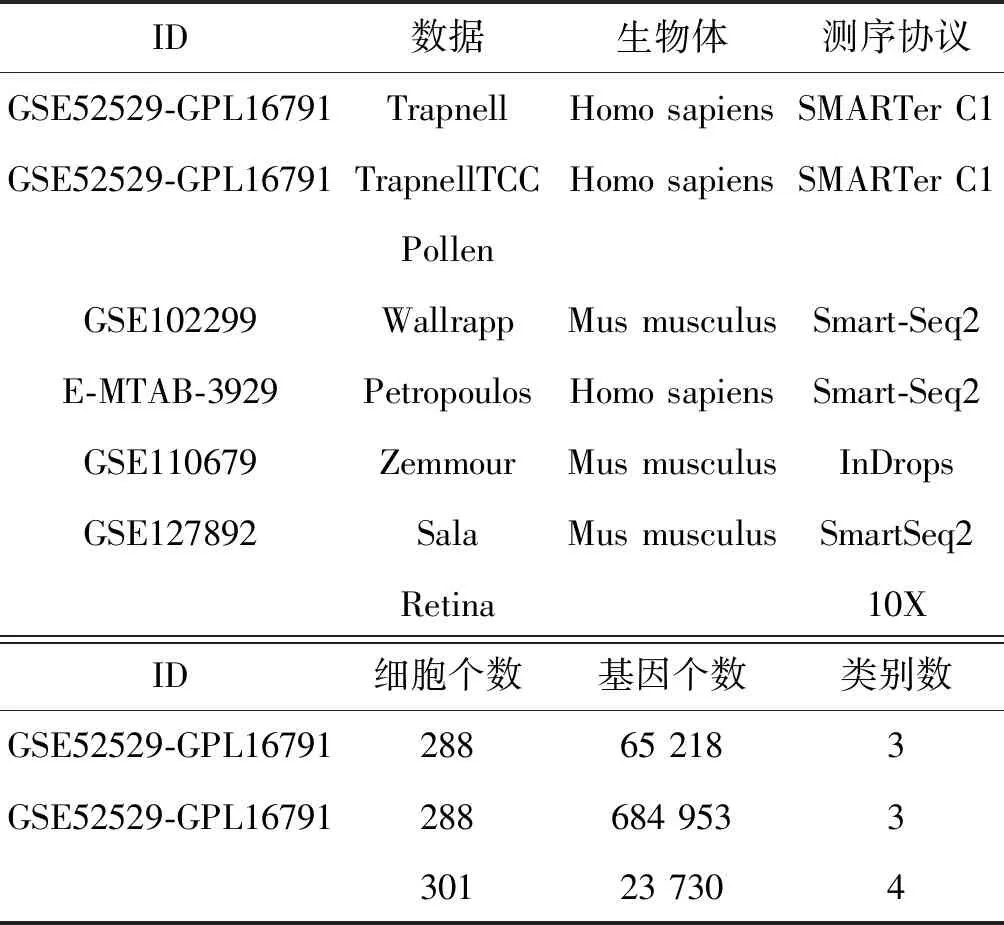
表1 多种数据类型scRNA-seq数据集
2.2 数据预处理
单细胞数据预处理已开发多种优秀程序包,如scater质量控制,Seurat选出高变异基因(HVG)。Scater实现删除低质量细胞及基因(零值、ERCC和pink-in),Seurat能选出高度变异的基因。这些预处理能使单细胞数据降低一定维度,但一般也接近上千维,接着用经典的PCA降维,scVI通过深度自编码器学习达到降维作用,对不同的数据集有一定效果。本实验为体现超图在单细胞数据上的表现效果,只使用Seurat提取高度变异基因,进行超图学习。
2.3 相似距离
细胞之间的距离是聚类的核心关键,欧氏距离被广泛应用于各分类和聚类方法中,但欧氏距离有一定的局限性,细胞聚类效果并不好。Pearson和Spearmam相关性对细胞分类或聚类效果较好,其范围在[-1,1]之间,值越大,相关性越高,与距离成反比。本次使用式(1)和式(2)计算细胞之间的Pearson和Spearman相似距离。单一距离具有一定的偶然性,本次将三者进行结合,对构造超图具有明显的有效性。
(1)
(2)
2.4 超图神经网络
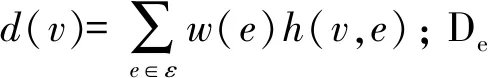
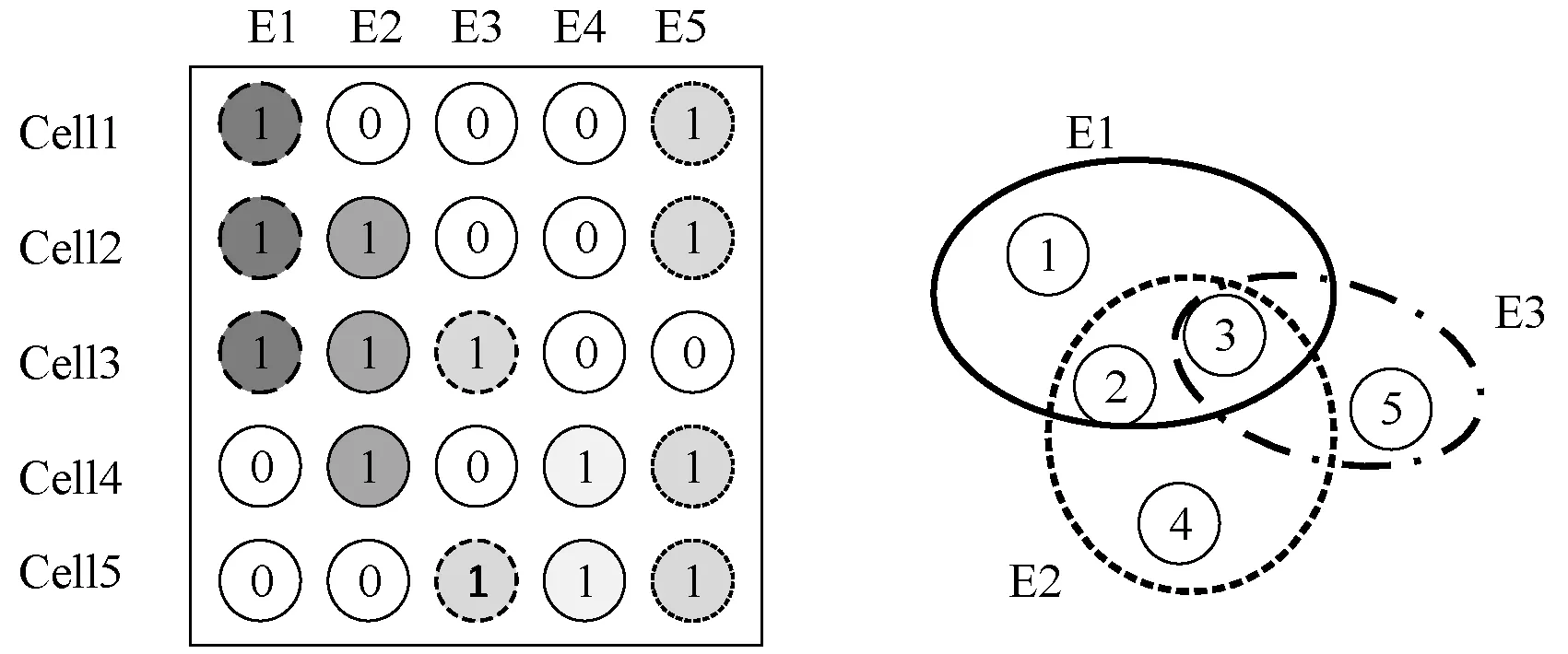
图2 细胞超图表示
(1) 构建顶点和边之间关系。
超图顶点和边之间的关系如式(3)所示,如果节点与节点之间相连接,用1表示,否则为0。
(3)
(2) 构建超图目标函数。
考虑超图每个顶点的下游分类问题,每个顶点的标签应该能够应用到超图结构中,整个学习的目标函数用式(4)描述。
(4)
式中:Remp(f)表示监督的损失;f(·)表示分类函数;Ω(f)表示超图的规范化,定义如式(5)所示。
(5)
(3) 通过超边卷积获取特征Y。
超图卷积由两个子模块组成:顶点卷积子模块和超边卷积子模块。顶点卷积将顶点特征集合到上边缘,然后上边缘卷积将相邻的上边缘特征集合到形心顶点。采用文献[15]的方法提取卷积后的特征如式(6)所示。
(6)
式中:H表示节点到边关联矩阵;X表示节点的特征;W、Θ代表学习参数。
(4) 构建超图神经网络分类器。
将多模态数据划分训练集和测试集,根据多模态数据之间复杂的相关性构建多个超边结构群,接着对超边群进行相连得到超边关联矩阵H,将关联矩阵H和节点特征输入到HGNN,得到节点输出标签。HGNN实现点到边再到点的特征转换,有效提取超图上的高阶相关性,通过多层不断学习经过Softmax得到预测标签。
2.5 多核超图的构建
在本文的基因对象分类任务中,N个可视对象数据的特征可以表示为X=[x1,x2,…,xn]T。本文建立超图根据两个特征之间欧几里得、Pearson和Spearman距离来计算d(xi,xj)。每个顶点代表一个细胞对象,每个超边是由一个顶点和它的K个最近邻构成,总共有N个超边,每个超边包含K+1顶点。因此,得到单个矩阵H∈RN×N,H中有N×(K+1)项等于1,其他的等于0;n个多核矩阵合并为H∈RN×nN。基因数据以图形结构组织,每个超边是通过连接一个顶点和它们的邻居节点来构建邻接关系。n个核得到n×N个超边和H∈RN×nN。
3 实验与结果分析
3.1 数据预处理
基因数据预处理,是分类准确的关键,大部分处理scRNA-seq数据的步骤包括删除少量异常细胞、删除表达值为0的基因、0值占比大于一定量的基因,这些操作对一些方法有一定效果。本实验直接对原始数据使用公认较好的Seurat方法选高度变异的基因。
具体步骤:创建Seurat对象(Create SeuratObject),不删除细胞和基因。
规范化处理如式(7)所示。
X=log(X+1)
(7)
式中:X是表示基因表达矩阵,X值可以是原始counts或FPKM、TPM值;X+1是为了防止表达值为0时取log出错。
根据离散度值(Dispersion)选出前10%基因(HVG),如图3所示。
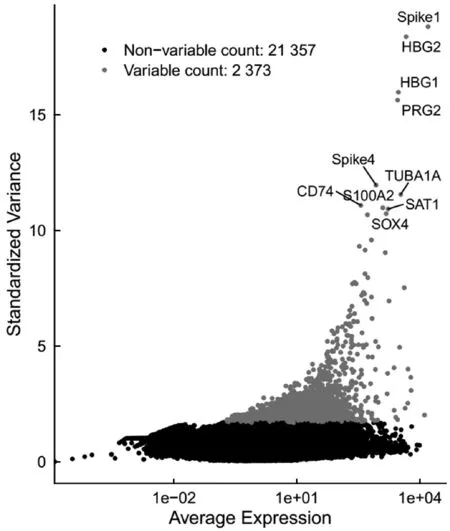
图3 高度变异基因
选用Pollen数据集,其中基因23 730个,选择高度变异的2 373个基因,如图3灰点所示。并标记出排名前十的基因,如Spike1、HBG2等基因。
3.2 超图神经网络学习
将基因数据集分两类,训练集70%,测试集30%。根据细胞两两之间的欧氏距离、Pearson和Spearman建立超图:其中每个顶点代表细胞,每个超边由细胞与细胞之间的K(K=10,15,20)个最近邻连接。将预处理数据直接输入超图神经网络进行学习,其中HGNN为两层,使用Softmax生成预测标签。
3.3 实验结果对比
实验使用不同测序技术平台产生的8个数据集进行验证,数据来源及描述见表1。其中scPred方法中,作者使用阈值为0.9能有较高准确率,但在本文数据集中,阈值设置0.9准确率较低。为提高准确率,本次实验降低阈值,设置为0.7。所有HGNN训练次数设置为600,学习率0.001。实验结果如表2所示。
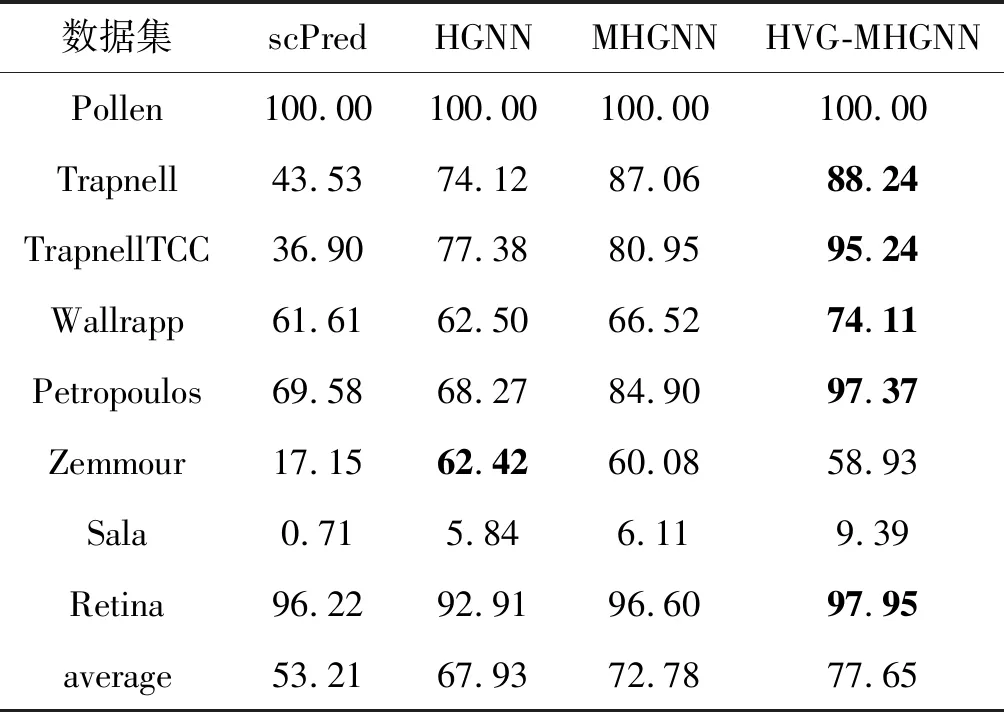
表2 多种算法在不同数据集上准确率的比较(%)
其中HGNN表示原始数据集直接经过HGNN处理;MHGNN为3种距离进行多核合并得出结果(其中Retina数据集较大,使用3种距离合并后进行超图学习,需占用大量内存,导致内存溢出,这里只选择欧氏距离和pearson距离);HVG-MHGNN是本文的方法,先选出高度变异的基因,再进行多核超图学习(其中Sala数据集准确率较低,可能不适合分类研究,或者其先验知识标注标签不对)。根据四个实验结果,HVG-MHGNN准确率在8个数据集中的5个最高,平均准确率最高。
实验平台采用WinServer 2019,处理器为E5-2620v4,2.10 GHz,内存为96 GB。多种算法在不同数据集上运行时间性能比较如表3所示,单位为秒。其中scPred使用R语言,HGNN采用Python的PyTorch。实验中由于使用语言及多距离合并方式不同。scPred只能与HGNN能进行实验对比,R语言在处理大型数据集时效率较低,Python读取数据及运行速度较快,较大数据集应选择Python处理更合理。MHGNN和HVG-MHGNN实验结果对比显示,MHGNN经过特征选择后(HVG-MHGNN)运行效率较高。

表3 多种算法在不同数据集上运行时间性能比较 单位:s
4 结 语
多种单细胞RNA测序技术为识别细胞类型和细胞异质性提供便利。本文提出一种基于特征选择(HVG)多核超图神经网络分类方法(HVG-MHGNN),对于超高维数据集,经过特征工程,然后根据多距离视角合并再进行HGNN特征表示学习对细胞分类准确率有较大提升。方法的合理性可从以下几点思考:(1) 主流的分类方法scPred已证明在生物学和临床场景中有很高准确性,但HVG-MHGNN与scPred对比,在相同数据集上分类准确率更高。(2) 多视角数据在单细胞测序成本上和采样技术上有一定限制。从单视角数据多距离的角度考虑单细胞测序数据聚类有一定的合理性,本实验结果已经证明MHGNN比HGNN准确率较高。(3) MHGNN在情感分析和链路预测已经有一定的应用场景,在单细胞测序数据上应用较少。
基于此,通过多种实验平台不同数据集证明了HVG-MHGNN方法的有效性。该方法虽在准确率有所提升,但在处理较大数据集时,超图学习需要大量内存,这也是图学习的一个缺点。从多视角收集单细胞数据是今后研究的一种主流趋势,这更能给临床带来诊断的参考意义。后续将继续研究超图如何在单细胞转录组数据集进行聚类,以及如何处理大型数据集。
猜你喜欢
新民周刊(2022年27期)2022-08-01
传染病信息(2021年6期)2021-02-12
科学(2020年4期)2020-11-26
无线电工程(2020年6期)2020-05-18
电脑爱好者(2018年2期)2018-01-31
生殖医学杂志(2015年11期)2015-02-28
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
电测与仪表(2014年6期)2014-04-04
四川电力技术(2014年2期)2014-03-19
