
非前药型第三代头孢菌素结构特征和谱学性质的计算分析
2023-10-09 10:22余德观陈旭雷翁约约廖颖艺王朝杰
光谱学与光谱分析 2023年10期
余德观,陈旭雷,翁约约,廖颖艺,王朝杰
1. 温州市人民医院药剂科,浙江 温州 325000 2. 温州市中心医院药剂科,浙江 温州 325000 3. 温州医科大学附属眼视光医院药剂科,浙江 温州 325003 4. 温州医科大学药学院,浙江 温州 325035
引 言
自20世纪80年代开发的大多数第三代头孢菌素因其抗菌谱广,组织分布好,不良反应较低等优点而在临床上广泛使用。但随着细菌耐药性的增加,尤以革兰阴性菌为主的多重耐药菌检出率呈快速上升趋势,产超广谱β-内酰胺酶(extended-spectrum β-lactamases,ESBLs)和碳青霉烯酶仍是肠杆菌科细菌最重要的耐药机制[1]。头孢菌素的结构是在母核——头孢菌素基本结构(basic structure of cephalosporin,BSC)基础上,通过改变其侧链结构提高体外稳定性,抗菌活性和对β-内酰胺酶的稳定性,如分别在α和α′位置增加氨基和氢原子,会导致碱性化合物在胃的酸性条件下质子化,7β氨基对抗菌活性(X=H)至关重要,用烷氧基(—OR)替换C-7处的氢可提高抗菌活性,当Z是五元杂环而不是六元杂环时,6α氢对于生物活性和抗菌活性是必需的[2]。因此,对于头孢菌素的结构研究是拓展其药学特性的有效手段。
光谱技术与量子化学计算相结合的方法已成为探究结构行为和观察药物分子电子结构最有效的方法之一[3]。有研究报道[4]X3LYP杂化密度泛函在预测谐波振动频率方面表现更准确,Kostjukova[5]等用X3LYP杂化密度泛函方法计算研究了吖啶橙染料在水溶液中的振动吸收光谱,分析了使用各种泛函和基组的计算结果后,证明其频率一致性最好。在头孢菌素的已有研究中,作为功能性药物载体的人血清白蛋白(human serum albumin,HSA),由于其具有较好的亲和性且三级结构、净电荷和溶解度大小均已熟知[6],Cheng等[7]研究了头孢克洛等头孢菌素与HSA的猝灭机制、结合模式和构象变化,并对其结合进行了热力学分析,证实了其与HSA之间存在相互作用,同时Dittrich[8]等用密度泛函理论进一步预测了头孢克洛的晶体结构和不同构象的静电势;Chaudhary[9]等通过光谱实验和理论量子化学方法研究了头孢拉定的振动光谱(红外和拉曼)、NBO、AIM、化学反应性等内容。
由于头孢菌素类品种繁多,其不同结构间的性质比较与特性仍缺乏系统性的研究。为了探索其内在性质,促进新药研发,确定药物结构变异的功能,针对我国抗感染药物医药市场中占据市场份额较大的第三代头孢菌素[10],筛选无需结构修饰,性质稳定的非前药型头孢菌素头孢布坦(ceftibuten,CTB)、头孢唑肟(ceftizoxime,CZX)、头孢地尼(cefdinir,CDR)、头孢克肟(cefixime,CEC)、头孢噻肟(cefotaxime,CTX)、头孢曲松(ceftriaxone,CRO)、头孢他啶(ceftazidime,CAZ)和头孢哌酮(cefoperazone,CFP)作为研究对象(见图1),将借助密度泛函理论(density functional theory,DFT)探索其结构与性质规律。
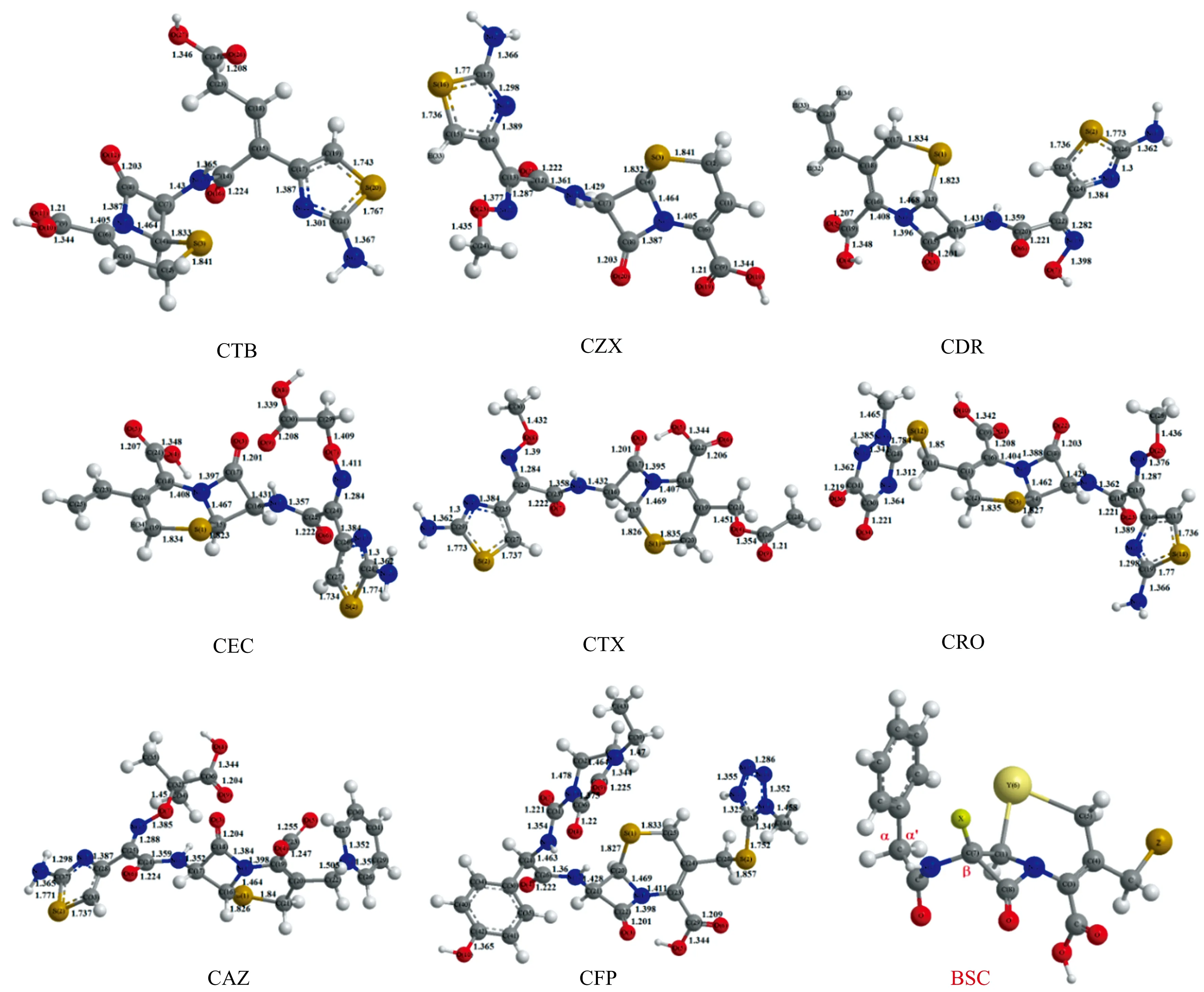
图1 非前药型第三代头孢菌素在X3LYP/6-311+G(d,p)水平下的主要键长及其母核结构Fig.1 The bond lengths and its core structure of the non-prodrug-type third-generation cephalosporins by X3LYP/6-311+G(d,p)
1 实验部分
1.1 几何结构优化、IR、UV谱计算
本研究采用密度泛函理论方法X3LYP,在6-311+G(d,p)基组水平上对非前药型第三代头孢菌素的几何结构进行优化,然后进行水相中的红外光谱、紫外-可见光谱计算,得到无虚频的能量极小结构,并对波函数进行电子结构分析。密度泛函计算利用Gaussian16[11]程序实现,Chemoffice2018绘制分子几何结构图,计算结果用Origin8.5作图,其中IR谱采用矫正因子(0.969 8)[12]并比较其计算和实验峰位归属。
1.2 分子表面静电势、分子前线轨道分析
将波函数文件在Multiwfn3.8[13]程序中分别对分子表面静电势、原子电荷、紫外吸收分子轨道进行分析,并通过可视化程序VMD和Multiwfn3.8绘制分子静电势分布图、轨道图和能隙数据。最后,根据可视化结果预测药物的活性反应位点,并对其光谱性质进行研究。
1.3 反应性描述符分析
利用概念密度泛函理论(conceptual density functional theory,CDFT)研究分子的亲电/亲核性,并解释其反应活性和反应位点。全局反应性描述符可用于阐明分子中的化学键、反应机理和反应位点,比较化合物的反应活性。全局反应性描述符包括:电离势(IP)、电子亲和势(EA)、电负性(χ)、整体硬度(η)、化学势(μ)、整体亲电指数(ω)和整体柔软度(S),计算公式如式(1)—式(7)
IP=-εHOMO
(1)
EA=-εLUMO
(2)
χ=-(εLUMO+εHOMO)/2
(3)
η=(εLUMO-εHOMO)/2
(4)
μ=-χ=(εLUMO+εHOMO)/2
(5)
ω=μ2/2η
(6)
S=1/η
(7)
在CDFT中引入福井函数,以衡量各个化合物当电子数或外部化学势发生变化时的电子密度差异。研究人员可以根据原子电荷,得出每个原子的福井函数值。一般来说,福井函数值越大,对应位点的反应性越大,f+(r)越大代表亲电性越大,f-(r)越大代表亲核性越大,f0(r)越大则表明其与自由基的反应能力强。简缩福井函数公式如式(8)—式(10)
亲核攻击:f+=q(N+1)-q(N)
(8)
亲电攻击:f-=q(N)-q(N-1)
(9)
自由基攻击:f0=[q(N+1)-q(N-1)]/2
(10)
式(8)—式(10)中:q为原子的电荷量;N为中性态;N-1为电离1个电子状态;N+1为结合1个电子状态。通过以上公式分别计算全局反应性描述符和局部反应性描述符参数,并列表分析其反应活性。
1.4 分子对接研究
通过从PDB蛋白质数据库下载人血清白蛋白(human serum albumin,HSA)靶向蛋白(PDB:1AO6),使用Pymol软件去除水分子和小配体后,加氢,计算电荷,导入POCASA系统(http://g6altair.sci.hokudai.ac.jp/g6/service/pocasa/)[14],并计算蛋白配体的最佳结合位点,用AutoDock软件进行分子对接,采用拉马克遗传算法(lamarckain genetic algorithm,LGA)进行对接运算。最后使用LigPlot+和Pymol对结果进行可视化分析。
2 结果与讨论
2.1 几何结构分析
分析非前药型第三代头孢菌素优化后的药物分子结构(图1),发现各头孢菌素基本结构BSC的二面角和键角相近,但由于取代基的不同,使得各个药物取代基附近的空间结构有所差异,其中N、O等原子对键长的影响较大。如CTB和CZX两个药物结构相近,对比两者之间的二面角数据发现,由于取代基的不同,CTB的C18-C15-C17-C19二面角为-33.697°,而CZX的N22-C13-C14-C15二面角却为-19.897°,相同结构间的二面角如C8-N5-C6-C9分别为55.474°和55.120°却非常接近;对比CDR和CEC两个药物,相同结构间的键角如CDR的 C22-N10-O7和CEC的C24-N12-O7非常相近,但由于取代基的不同,发现二面角中CDR的N10-C22-C24-C25和CEC的N12-C24-C26-C27刚好相反,这可能与取代基的空间效应有关,CEC的乙酸基占据体积较大,使噻唑环发生反转;对比CZX和CTX的几何数据发现,基本结构的键角数据也非常接近,由于CTX酯基的取代,C17-N10-C18-C22的二面角为45.998°,而CZX的C8-N5-C6-C9的二面角为55.120°,比较接近,由此可见该取代基对其他基团空间位置的影响不大。
由于非前药型第三代头孢菌素中已有的结构参数实验数据仅限CZX[15],对其的理论值和实验值进行线性回归(图2),计算决定系数R2和均方根误差(root mean square error,RMSE),对比实验和理论数据基本吻合。
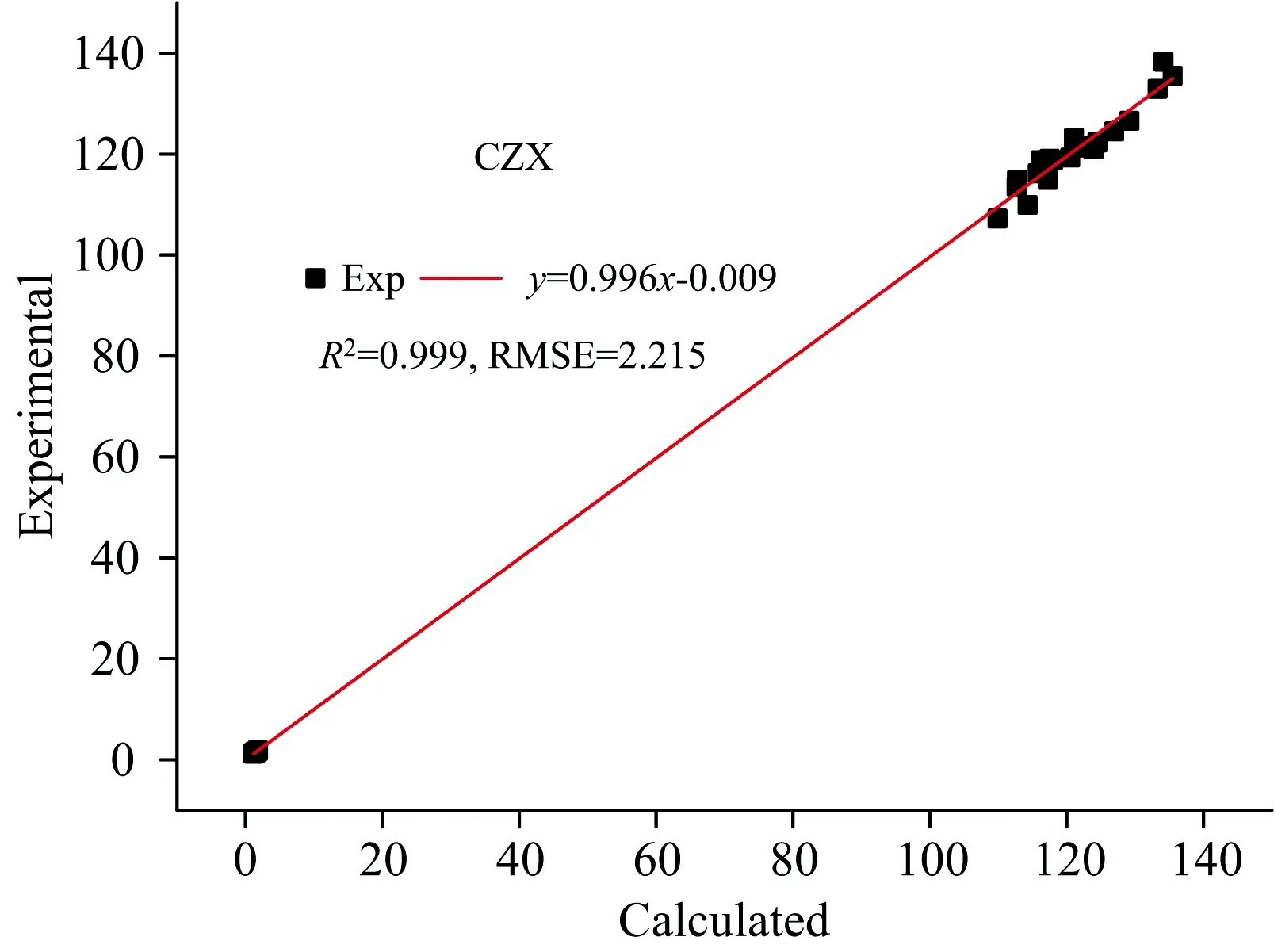
图2 在X3LYP/6-311+G(d,p)水平下CZX键长键角理论和实验值线性回归拟合Fig.2 The theoretically calculated bond lengths and bond angles of CZX by X3LYP/6-311+G(d,p) fit linearly with the experimental values
2.2 红外吸收光谱
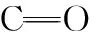
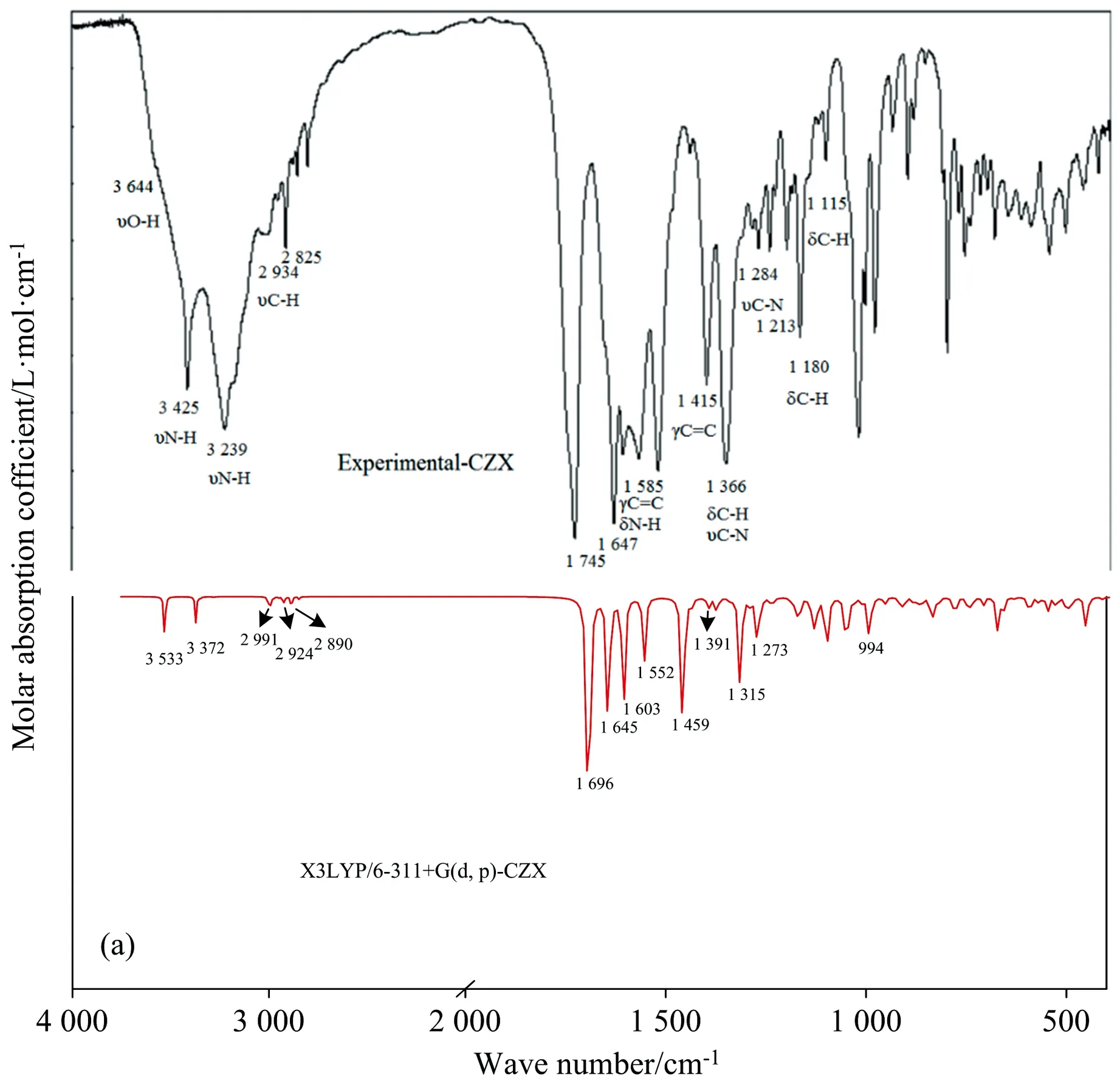
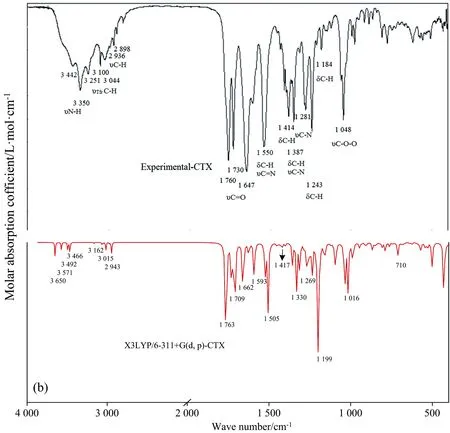
图3 CZX (a)和CTX (b) 的实验和理论光谱图Tfr:噻吩;ν:伸缩振动;γ:面外弯曲振动;δ:面内弯曲振动Fig.3 Experimental and calculated IR spectra of CZX (a) and CTX (b)Tfr:Thiofuran;ν:Stretching;γ:Out of plane bending;δ:In plane bending
以上图谱信息基本与实验吻合,但也有一些差别,这些差别主要因为理论计算模拟是计算单个分子的计算模式,而实验获得的结果是在固体粉末状下的分子共同表现结果,分子间存在多种相互作用造成的[17]。除此发现实验光谱3 330 cm-1附近出现较强水分峰[18],理论计算中却没有,这是由于计算单个分子的IR谱,所以没有氢键缔合的O—H峰。总体上,理论计算还是为分子的振动情况提供了较为准确的信息。


图4 水相中CTB至CFP在X3LYP/6-311+G(d,p)水平下的基态红外吸收光谱图Fig.4 Ground-state infrared absorption spectra of CTB to CFP in water by X3LYP/6-311+G(d,p)
2.3 紫外-可见吸收光谱
有机化合物的紫外吸收光谱可以反映分子结构中发色团和助色团的特性,通过光谱的解析可以了解共轭体系、空间位阻等对化合的影响。
非前药型第三代头孢菌素的紫外-可见吸收光谱在可见光范围内峰形较宽且强,最强紫外吸收峰主要集中200~250和300~350 nm范围[图5(a,b)]。其中,CTB、CZX、CDR、CEC、CTX和CRO的最强吸收峰分别出现在220、241、303、311、241和242 nm附近,对比CTB和CZX发现,由于CZX中N—O基团的吸电子能力比CTB中远端的羧基强,导致紫外波长发生红移;对比CZX和CTX发现,CTX由于酯基的存在使284 nm处出现一个强峰,原因为酯基的生色团作用,当酯基与BSC等基本结构处于非共轭状态时,总的吸收是各个生色团吸收的总和,表现结果为284 nm处吸收强度的显著增加;对比CDR和CEC发现,CEC在结构上由于乙酸基的存在,紫外峰整体发生了红移,这也充分说明了BSC母核的取代官能团存在吸电子基团时,能使紫外吸收峰发生红移。而CFP由于苯环的存在,紫外光谱中由π—π*跃迁产生的特征吸收带受到羟基取代基的影响,形成p—π共轭结构,吸收峰红移,吸收强度增大,精细结构消失,同时,由于BSC母核形成的共轭作用以及羰基生色团作用,降低了π*轨道能量,在吸收紫外光后可发生π—π*和n—π*跃迁,使吸收峰发生了红移。

图5 水相中在X3LYP/6-311+G(d,p)计算水平下非前药型第三代头孢菌素CTB-CEC(a)和CTX-CFP(b)的紫外光谱图Fig.5 UV-Visible spectra of the non-prodrug-type third-generation cephalosporins CTB-CEC (a) and CTX-CFP (b) in water by X3LYP/6-311+G(d,p)
通过计算的垂直激发能(E)、吸收波长(λ)、振子强度(f)和主要跃迁组态数据分析(表1)可知,其最大吸收峰的主要是由HOMO→LUMO+1,HOMO-2→LUMO和HOMO-1→LUMO+2等轨道电子跃迁贡献的。
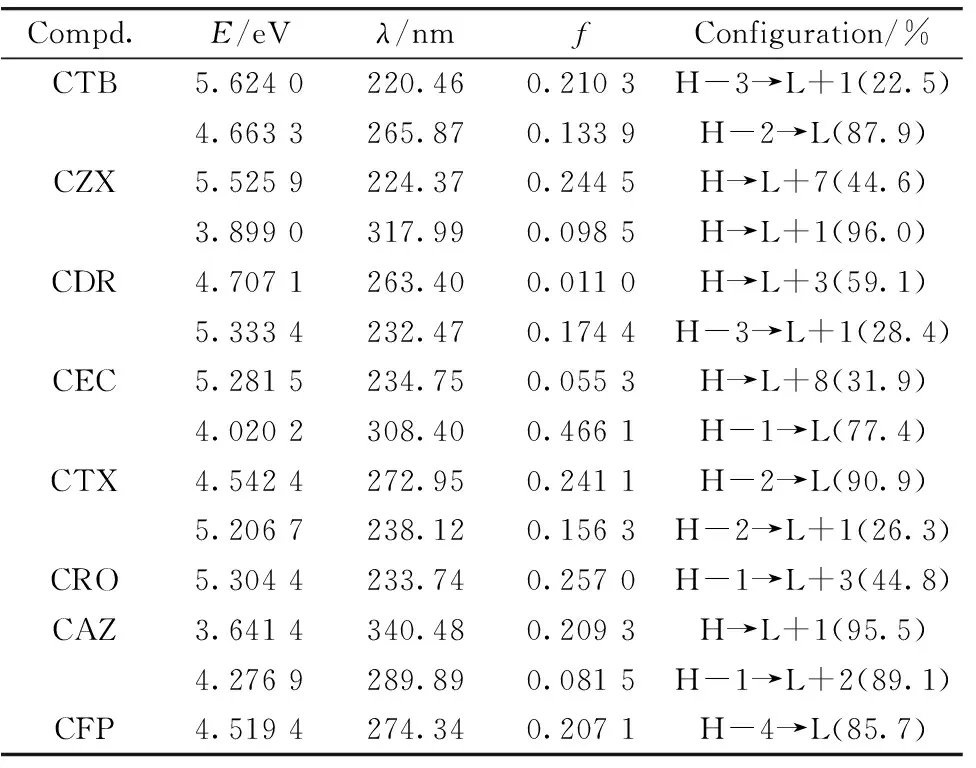
表1 水相中在X3LYP/6-311+G(d,p)计算水平下非前药型第三代头孢菌素的紫外-可见吸收光谱数据Table 1 UV-Visible data of the non-prodrug-type third-generation cephalosporins in water by X3LYP/6-311+G(d,p)
2.4 分子表面静电势
通过静电势图分析(图6),蓝色区域表示负电荷或亲电区域(电子的密集区域),易受到亲电试剂的进攻;红色区域表示正电荷或亲核区域,易受到亲核试剂的进攻;白色表示零电位区域。其中,黄球对应静电势极大点,蓝球对应静电势极小点。发现非前药型第三代头孢菌素中极大点大多分布在羟基和氨基的氢原子附近,而极小点主要分布在羰基O原子和N原子较集中的部位。如CTB的极大点主要分布在噻唑环以及其他部位在羧基中的羟基氢原子附近,极小点主要分布在羰基O原子附近,分析认为羟基在共轭条件下O原子的一对电子参与形成p—π共轭,使供电子能力较强,容易受到亲核试剂的攻击;而羰基等基团中,O的电负性比C大,诱导效应使之吸电子能力增强,容易受到亲电试剂的攻击。

图6 非前药型第三代头孢菌素在X3LYP/6-311+G(d,p)水平下的静电势等值面图Fig.6 Molecular electrostatic potential maps of the non-prodrug-type third-generation cephalosporins
2.5 前线分子轨道分析
前线分子轨道(frontier molecular orbital,FMO)是一种分子轨道理论,通过对前线分子轨道的计算分析,可以预测分子的得失电子能力,在分子附近,HOMO和LUMO轨道分别提供和接受电子。非前药型第三代头孢菌素的前线轨道能隙的范围都在3.62~4.49 eV之间(图7),CAZ分子相对于其他分子有较低的HOMO和LUMO能隙Δε=3.62 eV,说明该分子具有较高的占据轨道,供电子能力最强,分子最活泼。相反,CFP分子轨道的HOMO和LUMO的Δε较大,所受到的束缚力就较小,说明该分子更难接受电子。以上研究进一步描述了这些分子轨道间的电子离域性,从而解释了整个分子的稳定性。

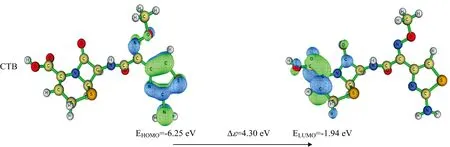

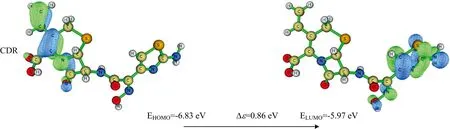

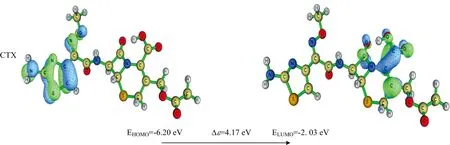

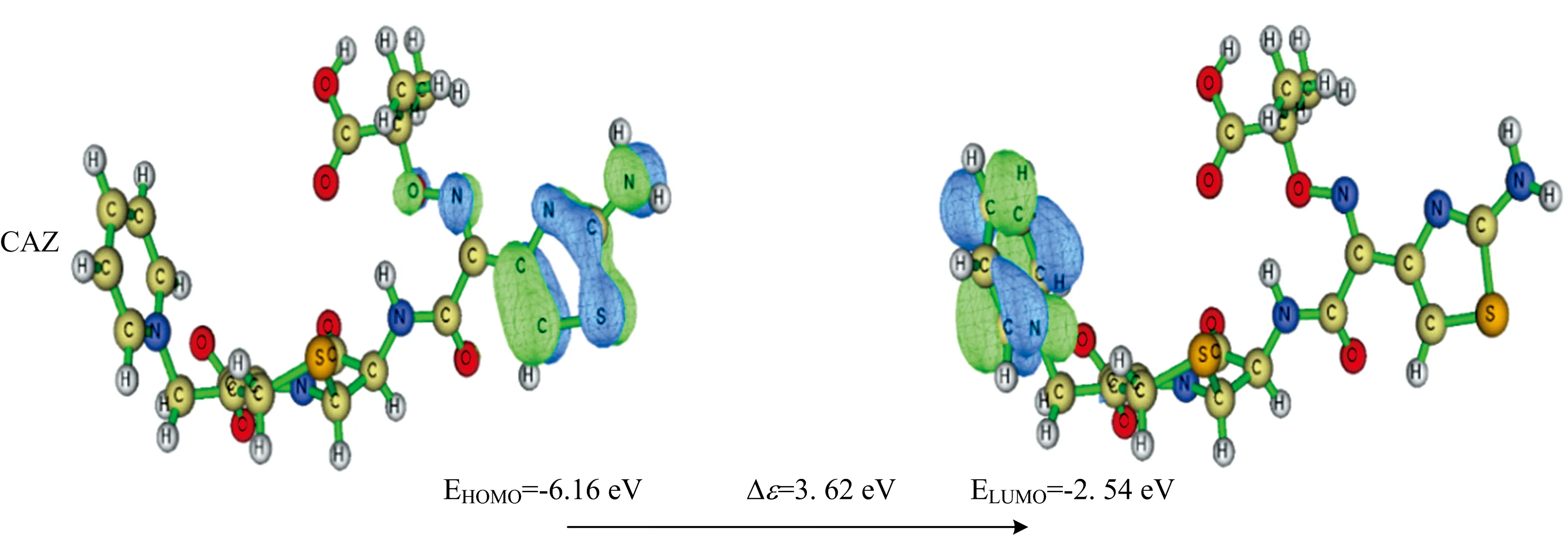

图7 非前药型第三代头孢菌素前线分子轨道图Fig.7 Frontier molecular orbital maps of the non-prodrug-type third-generation cephalosporins
2.6 反应性描述符
2.6.1 全局反应性描述符
通过分析全局反应性参数数据发现(表2),非前药型第三代头孢菌素之间的反应活性差异不大,其中,CAZ的亲电指数(ω)最大,表明其得电子能力强,反应活性最高,CFP的(μ)值最低,硬度(η)最大,说明其对微干扰下化学体系电子云畸变的抵抗能力较强,稳定性最好。

表2 非前药型第三代头孢菌素在X3LYP/6-311+G(d,p)水平下的全局反应性指数(单位:eV)Table 2 Global reactivity indexes of the non-prodrug-type third-generation cephalosporins by X3LYP/6-311+G(d,p) (Unit:eV)
2.6.2 局部反应性描述符
除了整体反应性参数外,分子中的原子的局部反应对于理解分子结构的反应机理也很重要。为进一步判断药物分子的活性位点,对非前药型第三代头孢菌素进行局部反应性指数分析,即简缩福井函数(condensed fukui function,CFF) 中的(f+,f-,f-)以及软度(s+,s-)和亲电指数(ω+,ω-)。分析非前药型第三代头孢菌素CFF的f-、s-和ω-数值发现(表3),CDR和CAZ的f-、s-和ω-数值最大,表明这两个药物分子的第2位S原子(噻唑环上)和CTB的第20位S原子(噻唑环上) 最容易受到亲电试剂的进攻;而分析CFF的f+、s+和ω+中的极大值发现,CAZ的f+、s+和ω+最大,表明该药物分子的第31位C原子(羧基的α碳)最容易受到亲核试剂的进攻。以上局部反应性描述符分析结果基本与静电势分析结果一致。
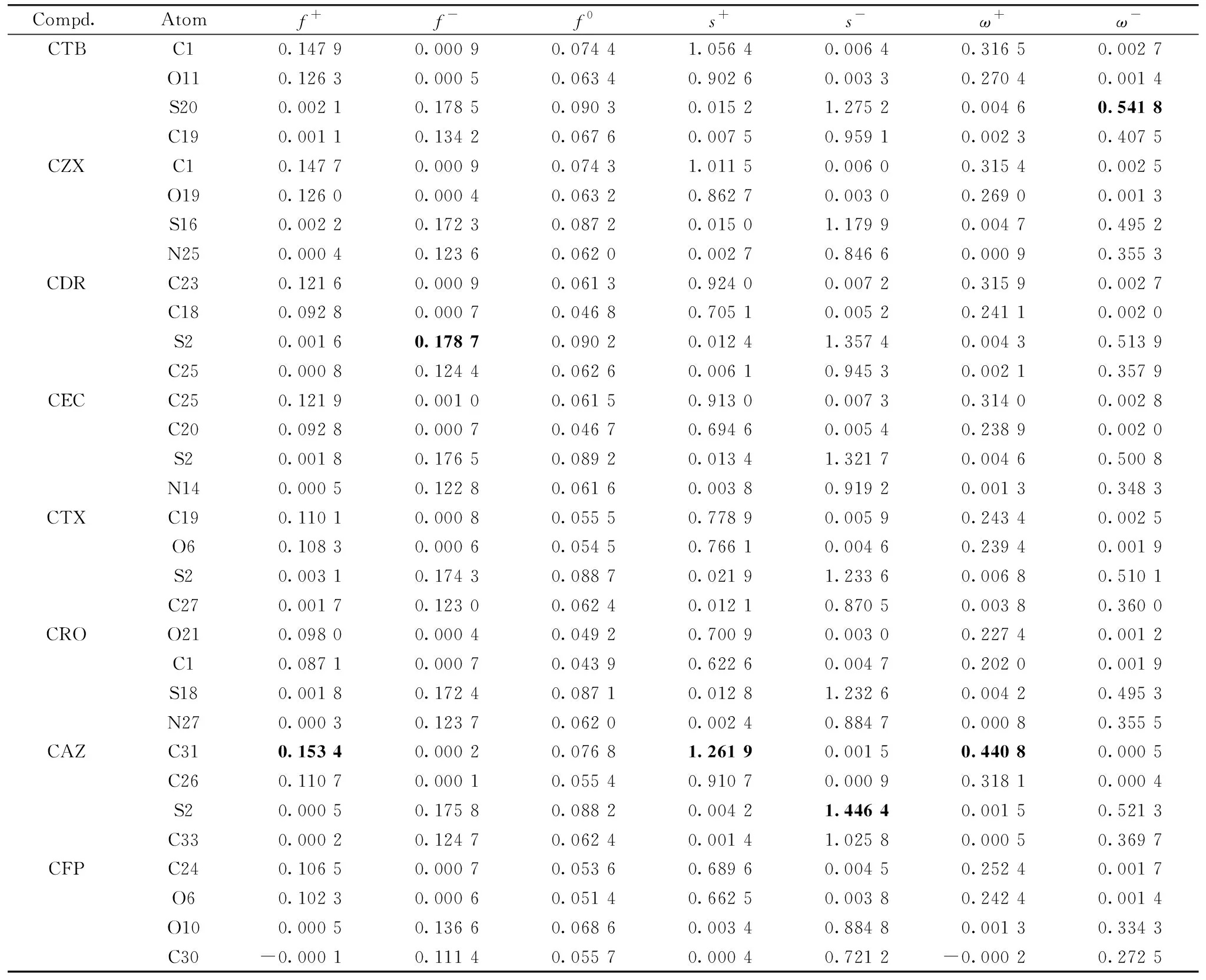
表3 非前药型第三代头孢菌素在X3LYP/6-311+G(d,p)水平下的简缩福井函数值(单位:eV)Table 3 The condensed fukui function value of the non-prodrug-type third-generation cephalosporins by X3LYP/6-311+G(d,p) (Unite:eV)
2.7 分子对接研究
分子对接可以对药物和生物大分子之间的相互作用做出模拟,可以更直观地了解药物与大分子之间相互作用的结合位点和作用力类型。通过分析CZX与HSA之间的结合方式发现(图8),其作用力以疏水作用力、氢键、π-阳离子相互作用为主,结合位点较深。非前药型第三代头孢菌素与HSA之间通过氢键作结合的残基主要有GLU292、ASN295、LYS444、ARG218、ARG222、ALA291等,通过疏水相互作用相结合的残基主要有TRP216、LEU238、HIS440等(图9)。发现形成氢键的原子大多为N或O原子,这与静电势的分析结果非常吻合,原因为极值点容易受到亲电或亲核试剂进攻,使该原子得到或者失去电子形成氢键。

图8 CZX与HSA的分子对接图Fig.8 Molecular docking diagram of CZX and HSA
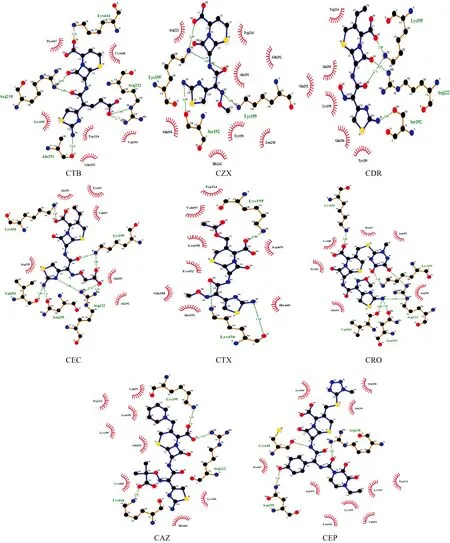
图9 非前药型第三代头孢菌素与HSA的结合模式图Fig.9 Binding mode diagrams between the non-prodrug-type third-generation cephalosporins and HSA
3 结 论
本研究系统全面地分析了非前药型第三代头孢菌素的结构性质,可为该类药物的生产质控、检测判断、新药开发、杂质鉴定等多方面提供参考。
猜你喜欢
广州化工(2022年19期)2022-11-09
广州化工(2022年18期)2022-10-22
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
硅酸盐通报(2020年1期)2020-02-25
猪业科学(2018年5期)2018-07-17
国外医药(抗生素分册)(2016年1期)2016-07-10
中国当代医药(2015年22期)2015-03-01
中国当代医药(2015年7期)2015-03-01
