
三克隆型多发性骨髓瘤临床特征及预后分析
2023-09-21 01:35陈瑜尤良顺陈志妹杨敏
浙江医学 2023年16期
陈瑜 尤良顺 陈志妹 杨敏
多发性骨髓瘤(multiple myeloma,MM)是一种克隆浆细胞异常增殖的恶性疾病,约占血液系统恶性肿瘤的10%[1],是血液系统第2 位常见恶性肿瘤[2]。MM 临床表现多样,常导致患者多器官损害,出现骨质破坏、贫血、肾功能不全、高钙血症,且易发生感染等[3]。MM好发于中老年人,目前仍无法治愈[4],尤其是高危类型的MM 患者疾病进展快,预后差[5]。根据患者临床表现、诊断、治疗及预后的不同,MM 可分为典型MM 和变异型MM。变异型MM 包括孤立性浆细胞瘤、髓外浆细胞瘤、冒烟型骨髓瘤、不分泌型骨髓瘤、骨硬化型骨髓瘤、双克隆及三克隆型骨髓瘤。其中双克隆及三克隆型MM 为极罕见类型,前者在MM 中的发生率为0.5%~2.5%,后者仅有个例报道[6]。本研究报道近年来浙江大学医学院附属第一医院确诊的2 例三克隆型MM,分析三克隆型MM 的实验室特点、临床特征及预后情况,为临床识别和诊治罕见类型疾病提供参考。
1 资料和方法
1.1 一般资料 回顾性分析2020 年9 月30 日至2023年3 月20 日在浙江大学医学院附属第一医院检测M蛋白的26 843 份标本。所有检出病例诊断均符合国际骨髓瘤工作组的诊断标准,经免疫固定电泳(immunofixation electrophoresis,IFE)分型确诊为三克隆型MM。
1.2 方法 检出病例均进行血常规、血生化、免疫球蛋白全套、IFE、骨髓细胞形态学检查、流式免疫分型、细胞因子检测、染色体检查、荧光原位杂交(fluorescence in situ hybridization,FISH)检测。分析三克隆型MM 患者的实验室资料、临床特征及预后情况。
2 结果
2.1 三克隆型MM 的检出率 26 843 份IFE 标本中,共检出M蛋白阳性9 197份,占34.26%;确诊MM 5 966例,占M 蛋白阳性标本的64.68%;双克隆型MM 109 例,占MM 标本的1.83%,三克隆型MM 仅2 例,占MM 标本的0.03%。
2.2 三克隆型MM 患者临床资料分析
2.2.1 基本情况 例1为男性,年龄69岁;例2为女性,年龄81 岁。初诊时均有贫血、肾功能损伤,肌酐、CRP、ESR、IL-6 均升高,例1 血β2 微球蛋白升高,例2 未检测。免疫球蛋白检测显示IgG 均升高,IgM 均降低,血清中轻链κ和轻链λ均升高,血清游离轻链κ/λ均异常。
2.2.2 骨髓细胞学检查和流式免疫分型 例1 骨髓细胞学检查结果显示:浆细胞明显增生,原始幼稚浆细胞占68.0%,成熟浆细胞占2.0%,见图1A;流式免疫分型结果显示:异常浆细胞群约占非红系细胞的53.27%,表达CD38、CD138、CD19(部分)、CD28、CD20、轻链λ,不表达CD56、CD27、CD117、轻链κ,见图1B。例2 骨髓细胞学检查结果显示:浆细胞增生活跃,原始幼稚浆细胞占4.5%,成熟浆细胞占5.0%,见图2A;流式免疫分型结果显示:异常浆细胞群约占非红系细胞的1.72%,表达CD38、CD138、CD56、CD117、B 细胞成熟抗原、轻链λ、轻链κ,不表达CD19、CD27、CD28、CD20,见图2B。
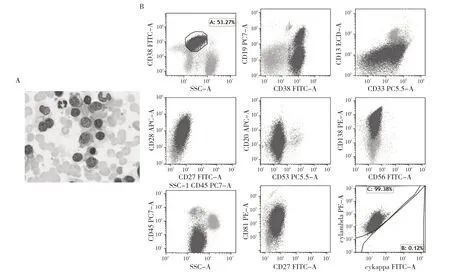
图1 例1 的骨髓形态学和流式免疫分型(A:骨髓形态学检测;B:流式免疫分型检测)

图2 例2 的骨髓形态学和流式免疫分型(A:骨髓形态学检测;B:流式免疫分型检测)
2.2.3 FISH 检测和染色体检查 例1 的FISH 结果显示:免疫球蛋白重链基因(immunoglobulin heavy chain,IGH)基因(14q32)分离重排50%,1q21 扩增60%,D13S319 基因(13q14.3)缺失50%,p53 基因(17p13.1)未缺失,RB1 基因(13q14)缺失50%;染色体检查核型结果显示:46XY[20],为正常核型结果,见图3A。例2的FISH 结果显示:1p32/CDKN2C(cyclin-dependent kinase inhibitor 2C)基因未缺失,IGH 基因(14q32)未分离重排,1q21 未扩增,D13S319 基因(13q14.3)未缺失,p53 基因未(17p13.1)缺失;染色体检查核型结果显示:46,XX,-3,-5,+2mar[5]/46,XX[5],为染色体异常核型结果,本次检查共分析10 个分裂象,除5 个为正常女性核型外,出现两条标记染色体,同时缺失1 条3 号染色体和1 条5 号染色体,见图3B。

图3 染色体核型分析(A:例1;B:例2)
2.2.4 IFE 检测 普通IFE 和经β 巯基乙醇解聚IFE 结果显示:2 例患者均具有3 种M 蛋白。例1 为IgG-λ+λ+λ 三克隆型,其中IgG-λ 和λ 条带致密且清晰,另外1 条λ 条带相对较弱;随着疾病进展,该条λ 条带逐渐加深,见图4。例2 为IgG-λ+λ+IgA-K 三克隆型,其中IgG-λ 和λ 条带致密且清晰,IgA-K 中K 条带相对较弱,见图5。

图4 例1 的IFE 结果(A:2020 年9 月21 日普通IFE;B:2020 年9 月21 日经β 巯基乙醇解聚IFE;C:2021 年6 月30 日经β 巯基乙醇解聚IFE;D:2022 年2 月7 日经β 巯基乙醇解聚IFE)

图5 例2 的IFE 结果(A:普通IFE;B:经β 巯基乙醇解聚IFE)
2.3 诊断 例1 诊断为IgG-λ+λ+λ 三克隆型MM,Durie-Salmon(DS)分期Ⅲ期,国际分期系统International Staging System(ISS)分期Ⅲ期;例2 诊断为IgG-λ+λ+IgA-K 三克隆型MM,DS 分期Ⅲ期。
2.4 治疗及预后 (1)例1:因体检发现血肌酐升高就诊,于2020 年9 月30 日开始用VRD(硼替佐米2.3 mg d1、d8、d15、d22+来那度胺25.0 mg d1~10+地塞米松40.0 mg d1、d8、d15、d22)方案治疗。两个疗程后评估疗效为部分缓解。患者VRD 第3 疗程治疗后,复查M蛋白百分比24.3%,M 蛋白含量19.2 g/L;骨髓涂片检查结果显示:原始+幼稚浆细胞样瘤细胞占42.0%。患者贫血进行性加重,考虑疾病进展。2021 年6 月4 日开始应用PCD(硼替佐米2.2 mg d1、d8、d15、d22+环磷酰胺0.5 g d1、d8、d15、d22+地塞米松40.0 mg d1、d8、d15、d22)方案,1 个疗程后复查血常规:Hb 49 g/L,PLT 47×109/L;骨髓涂片结果显示:原始浆细胞占42.0%,成熟浆细胞占5.0%,考虑患者疾病再次进展,2021 年9 月1 日予以VPD(泊马度胺4.0 mg d1~21+硼替佐米2.3 mg d1、d8、d15、d22+地塞米松20.0 mg,d1~2、d8~9、d15~16、d22~23)方案治疗。由于患者骨髓抑制较明显,2021 年10 月8 日改泊马度胺4.0 mg d1~14+硼替佐米2.1 mg d1、d8、d15、d22+地塞米松片750 μg/片,26 片,d1~2、d8~9、d15~16、d22~23。2022 年2 月6 日,患者出现胸闷气急,于2022 年2 月9 日入住本院ICU,需要血液透析。考虑患者MM 累及多个器官系统,肾功能衰竭,呼吸衰竭,感染加重,循环衰竭,生命体征不稳,家属放弃治疗。(2)例2,因间歇双下肢水肿就 诊,于2023 年1 月30 日给予PCD 方案(硼替佐米1.8 mg d1、d4+环磷酰胺0.4 g d1)化疗1 个疗程,同时止吐对症支持治疗,过程顺利。2023 年2 月15 日开始予以伊莎佐米联合地塞米松口服治疗。2023 年3 月3日因肾功能不全在肾脏病中心病房治疗,2023 年3 月10 日诊断肾衰竭。与患者家属沟通病情,告知目前病情危重,出血风险高,随时可能出现颅内出血、消化道出血、气道出血、呼吸心跳骤停等情况,家属放弃治疗。
3 讨论
三克隆型MM 的诊断需符合MM 的诊断标准,并经IFE 确认存在3 组M 蛋白。IFE 是M 蛋白鉴定的金标准[7],其特异度和灵敏度也保证了对三克隆型MM 临床诊断的重要意义。IFE 检测时M 蛋白多以单体形态出现,但也有少量聚合体形式存在[8]。本研究2 例患者IFE 结果均经β 巯基乙醇解聚IFE 确定,避免了因聚合体而造成的假阳性结果[9]。双克隆和三克隆型MM 患者的血清中含有2 或3 种M 蛋白[10],在IFE 时出现2 个或3 个不同的单克隆免疫球蛋白条带。许多研究显示MM 具有不同的克隆进化模式,其基因组的高度不稳定导致了克隆内部异质性[11-12],因此MM 患者体内存在不同的克隆亚群。目前认为双克隆型MM 的2 种单克隆可能来源于1 群恶变浆细胞,也可能分别来源于2群恶变浆细胞[13]。鉴于其罕见性,国内外暂无对双克隆/三克隆型MM 大宗病例报道[14],其产生机制尚未完全清楚。本研究中,2 例三克隆型MM 患者经IEF 检测发现有较强M 条带和较弱M 条带。一般患者经过治疗后,通常会出现1 种M 蛋白成分减少或消失[15]。但本研究中,并未出现某种M 蛋白成分的减少。随着疾病的进展,1 例IgG-λ+λ+λ 三克隆型MM 患者的第3 条λ 轻链逐渐加深,提示M 蛋白成分含量在逐渐增加中,可能与存在恶性克隆相关[16]。患者治疗效果不佳,这与典型的MM 及双克隆型MM 的治疗效果差异大。随着分子生物学及二代测序技术的发展,MM 克隆异质性和克隆演变的基因表达谱亦可提供额外的预后价值[17]。Soekojo 等[18]提出了功能性高危MM 患者的基因组学特征为预后较差、IL-6/JAK/STAT3 通路的突变增加、伴有异常有丝分裂和DNA 损伤反应相关的基因表达谱异常。本研究的2 例患者体内均存在3 个克隆亚型,IL-6 均升高,可能存在基因表达谱异常。但目前克隆演变研究大都处于科研阶段,因而临床基因检测比较困难。
本研究中2 例患者初诊时都存在贫血、肾功能损伤、骨病等相关表现,具有一般MM 的典型症状,但治疗效果不佳,发生早期器官衰竭,这可能与高危遗传学异常有关。病例1 FISH 检测结果显示IGH(14q32)分离重排,1q21 扩增,D13S319(13q14.3)和RB1(13q14)缺失。根据Mayo 骨髓瘤分层及风险调整治疗分层系统[19],此病例符合“双打击”骨髓瘤特征。Walker 等[20]研究结果显示,“双打击”患者中位总生存期(overall survival,OS)20.7 个月,中位无进展生存期(progression free survival,PFS)15.4 个月,属于极高危。马东升等[21]研究也提示“双打击”MM 的预后较R-ISS 分期Ⅲ期的患者更差,为极高危群体。例1 和例2 的染色体检查中都出现亚二倍体,是MM 预后不佳的独立指标。Pawlyn 等[22]研究提示亚二倍体是骨髓瘤中基因组最不稳定的亚群,而且还与高危性相关。病例2 染色体核型结果出现两条标记染色体,同时缺失1 条3 号染色体和1 条5 号染色体,推测和免疫球蛋白恶性克隆性增生相关,是患者预后较差的原因之一[23]。另有研究显示伴有染色体异常的MM 预后差[24]。本研究例1 OS为18 个 月,例2 为3 个 月,2 例患 者中 位OS 为10.5 个月。这与Yang 等[25]的研究中新诊断MM 的中位PFS 和OS 分别为41.6 和71.5 个月有比较大的区别,原因可能是本研究2 例三克隆型MM 都是极高危类型,疾病进展快,预后极差,因而生存时间短。
综上所述,三克隆型MM 临床极罕见,发病机制尚不完全清楚,无明显临床特征,诊断除MM 标准外,需IFE 证实3 种M 蛋白的存在。本研究中三克隆型MM疾病进展快,治疗棘手,患者易发生早期器官衰竭和死亡,预后极差,属于极高危类型,提示临床医生须高度重视。
猜你喜欢
安徽医科大学学报(2022年6期)2022-07-13
现代临床医学(2022年1期)2022-02-12
中国临床医学影像杂志(2021年5期)2021-08-13
天津医科大学学报(2021年3期)2021-07-21
中华养生保健(2020年7期)2020-11-16
武警医学(2018年10期)2018-11-06
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
实用皮肤病学杂志(2015年4期)2015-12-22
中国药理学与毒理学杂志(2015年3期)2015-12-16
郑州大学学报(医学版)(2015年2期)2015-02-27
