
嵌合抗原受体T 细胞免疫疗法追溯与展望
2023-09-20 12:34伍学强
中国现代医药杂志 2023年8期
伍学强
嵌合抗原受体T 细胞免疫疗法(Chimeric antigen receptor T-Cell immunotherapy,CAR-T),是抗肿瘤治疗最有发展前景的技术之一,目前已成为治疗非霍奇金淋巴瘤和急性淋巴细胞白血病的研究热点。从T 细胞的发现到转基因技术的发展进步,从CAR-T 技术的产生到临床应用,从在淋巴瘤、急性白血病等血液肿瘤中的应用到实体瘤的治疗,CAR-T 技术逐步成熟。本研究通过对CAR-T 技术发展历程的回顾,聚焦问题与挑战,探讨CAR-T 进一步发展的方向,以期造福更多肿瘤患者。
1 T 细胞的认识过程
1.1 T 细胞的发现1961 年,法裔澳大利亚科学家雅克·米勒(Jacques Miller)[1]领导完成了四项开创性研究,包括T 细胞的鉴定、T 细胞的选择以及T细胞与其他免疫细胞的相互作用,并确定了胸腺并不是一个退化的器官,而是免疫细胞发育的关键部位,对于机体所需的复杂而微妙的免疫系统而言,胸腺是不可或缺的。1970 年,Cerottini、Nordin 和Brunner[2]等人证明,细胞毒性活性来自于胸腺来源的T 淋巴细胞。进一步研究发现,T 细胞不仅有细胞毒性活性,更具有免疫记忆,即在获得性免疫中,机体对同一种抗原的再次出现,反应会更强烈。1971 年,Miller[3]和Gershon[4]研究小组分别通过不同的方法,独立证明了T 细胞免疫记忆的存在。同时发现,T 细胞介导免疫反应,具有致病性。至20世纪70 年代,T 细胞被证实对自身免疫性疾病的启动至关重要。Clagett 和Weigle[5]在一项早期研究中,用小鼠证明了实验性自身免疫性甲状腺炎的同种异体转移需要T 细胞的存在。同时,研究者发现T 细胞在移植免疫和器官排斥反应中具有重要功能,这一发现是定义T 细胞异体识别途径的关键基石。
1.2 T 细胞的分类
1.2.1 T 细胞亚群 早期研究发现,整个T 细胞群体可以被分为两个具有不同功能和表型的亚群,后来被称为CD4 和CD8 分子,分别标记这两个主要具有辅助性功能和细胞毒性功能的T 细胞群体。1986 年,研究者[6]进一步发现CD4+T 细胞群体由一系列不同的亚群组成,并且这些亚群具有多种多样的重要功能。这一研究证明了辅助性T 细胞(T helper cells,Th)的存在和不同特征,比如Th1 及Th2,这也是Th 细胞谱系异质性研究的肇始。2005年发表的两篇论文[7,8],鉴定并描述了一种新的产生IL-17 的CD4+辅助性T 细胞谱系:Th17 细胞。这一发现及相关研究,澄清了这种独特T 细胞亚群的分化过程和生物学特征。辅助性T 细胞主要为CD4+T 细胞,初始CD4+T 细胞(Th0)在各种抗原等的刺激下分化成包括Th1、Th2、Th17 在内的多种Th。细胞毒性T 淋巴细胞(Cytotoxic lymphocyte,CTL)主要指CD8+T 细胞,主要在受损的体细胞中,通过细胞毒性蛋白触发死亡途径。
调节性T 细胞(Regulatory T cell,Treg)是T 细胞的独特群体,可在抑制免疫反应中起作用。CD4+调节性T 细胞主要包括自然调节性T 细胞(Naturally occurring regulatory T cell,nTreg)及适应性调节性T 细胞(Adaptive regulatory T cell,aTreg),特征为高表 达CD25及转录因子FoxP3。1995 年,Shimon Sakaguchi[9]等发现,CD25 作为一种调节性T 细胞表面标记物,可以用来分离Treg。此外,Treg 还包括CD8+Treg,也与肿瘤及免疫疾病的发生发展密切相关。见图1。
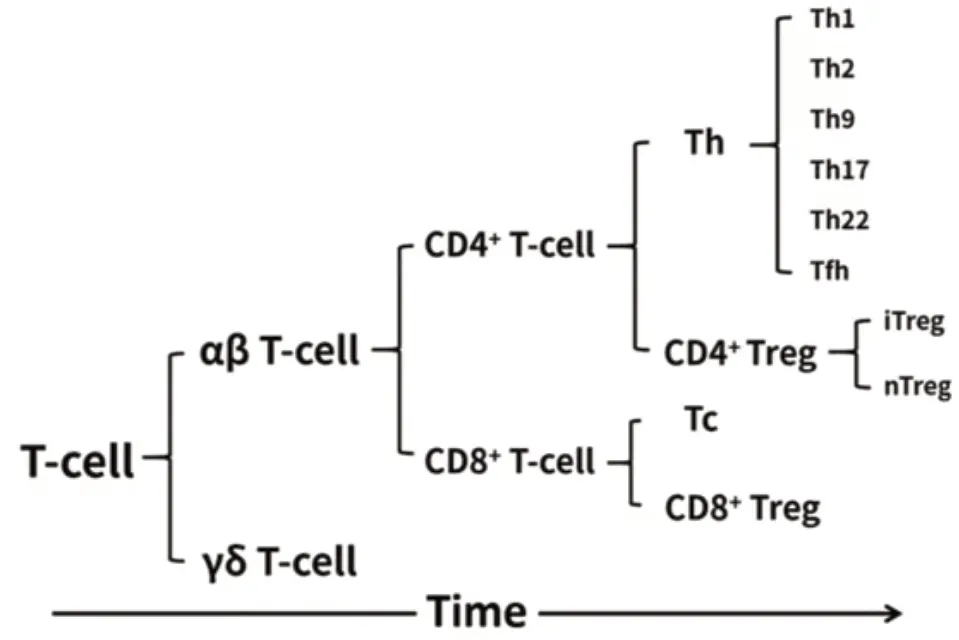
图1 T 细胞的分类
1.2.2 T 细胞受体(T cell receptor,TCR)分型 TCR为所有T 细胞的特征性标志,主要包括两种类型,分别是αβ 链受体和γδ 链受体。α 亚基和β亚基连结构成αβ 链,这一类T 细胞被称为αβ T 细胞,即我们通常所说的T 细胞,大约占95%。γδ T 细胞中由γ 亚基和δ 亚基连结成γδ 链,与αβ T 细胞不同,它们不仅表达不同的T 细胞受体(TCR),在生物学特性和活化模式方面也各不相同。γδ T 细胞谱系的发现、其TCR 的检测和描述、以及对其初始特征的确定,都是T 细胞研究领域非常重要的成就。
1.3 T 细胞作用方式T 细胞受抗原刺激进而分化,发挥作用,抗原的识别主要通过主要组织相容性复合体(Major histocompatibility complex,MHC)进行,其为主要决定免疫排斥反应的基因簇,分为Ⅰ类和Ⅱ类。1974 年,Doherty 和Zinkernagel[10]首次描述了MHC 限制的现象,他们研究发现,CTL 只识别病毒感染的靶细胞上的MHC-I 分子,认为T 细胞主要通过TCR 特异性识别和结合蛋白质抗原,其在识别抗原递呈细胞传递的抗原信息时,必须通过先识别递呈细胞上的分子类型。进一步研究证明,Th细胞识别MHC-Ⅱ类分子。但1994 年研究者发现恒定自然杀伤T 细胞(invariant natural killer T cells,iNKT)[11],违背了上述规则,由非MHC 递呈非肽抗原给T 细胞,进而研究者对T 细胞如何识别产生了兴趣。20 世纪80、90 年代的一系列突破性研究,让人们得以鉴定、克隆并测序了T 细胞上的抗原结合受体。T 细胞通过表达的独特粘附分子和趋化因子受体,“归巢”到不同的淋巴组织或外周组织。
1986 年,Martin[12]等人和Weiss[13]等人分别独立发现了激动型抗体刺激CD28(当时称为Tp44)会导致T 细胞激活信号的显著放大,但前提是T 细胞通过TCR 复合物受到了抗原的预先刺激。即T细胞识别抗原之后,仍需要第二个信号才能完全发挥其效应功能。T 细胞的完全活化依赖TCR 提供的抗原刺激信号和共刺激受体提供的共刺激信号。CD28 是T 细胞的一种表面分子,也是第一个被发现的T 细胞共刺激受体。T 细胞被抗原激活后会分化为相应的效应T 细胞及记忆T 细胞。效应T 细胞主要合成、表达并分泌各种淋巴因子而发挥效应,同时少部分被保留成为较长久存在的记忆细胞。
1.4 T 细胞耗竭1998 年,由Rafi Ahmed[14]和Rolf Zinkernagel[15]领导的两个研究小组研究了T 细胞对小鼠慢性淋巴细胞性脉络脑膜炎病毒感染的反应。他们使用四聚体染色检测了病毒特异性CD8+T 细胞,并在慢性感染期间检测到一群功能无反应的细胞,称这一现象为T 细胞耗竭。随后的研究证实,耗竭T 细胞在人的慢性感染中普遍存在,也证实了耗竭T 细胞存在于癌症中。此后,进一步的研究描述了T 细胞耗竭的免疫生物学特征,有望帮助我们通过逆转 T 细胞耗竭来恢复T 细胞的功能。
2 CAR-T 技术的产生及发展
2.1 嵌合抗原受体(Chimeric antigen receptor,CAR)CAR-T 技术的实现归因于转基因技术的发展。CAR 是一种基因编辑后的人工跨膜受体,将多个不同受体的特定结构域合而为一从而可以识别特定的抗原。T 细胞杀伤靶细胞主要依赖于TCR 识别靶细胞上MHC-I 递呈的特异性多肽。然而,实体肿瘤中MHC-I 大多是下调的,这就导致T 细胞无法特异性识别肿瘤细胞。由此,识别靶细胞的CAR成为CAR-T 技术的的关键之处。根据CAR 结构域的功能差异,可将CAR 分成三个功能区域,即:抗原识别区、跨膜区和胞内信号转导区[16]。
临床试验中,研究者从患者外周血中采集、分离出T 细胞,然后在实验室对它们进行基因修饰,将编码CAR 的基因导入[17],从而这些T 细胞可以表达此种受体。对这些经过基因修饰的T 细胞增殖后,将其回输到患者体内,这些T 细胞通过其表达的CAR 受体与靶细胞结合,从而触发一种内部信号产生,接着这种内部信号强效地激活这些T 细胞,以致使它们快速地摧毁靶细胞。
2.2 CAR-T 临床应用2008 年Fred Hutchison 肿瘤研究所首次将CAR-T 技术用于治疗B 细胞淋巴瘤,虽然疗效不明显,但是证实了CAR-T 技术的安全性。2010 年CAR-T 首次成功使一例B 细胞淋巴瘤患者病情得到控制,证实了其有效性。2011 年,美国Carl June 教授团队[18]将CAR-T 用于治疗B 细胞来源的慢性淋巴细胞白血病,达到CR,此为一大突破。
随后大量基于CAR-T 细胞的临床试验逐步展开。至今CAR-T 经历至少三代的技术迭代,主要差别在于胞内信号转导结构域的从无到有、从少到多地增加了T 细胞共刺激结构域,增强了T 细胞的杀伤功能和增殖活性,弥补了非MHC-I 依赖性激活时缺少共刺激信号的问题。目前临床批准的CAR-T 产品以第二代为主,部分三代CAR-T[19]产品已进入临床试验。
自2017 年两款CAR-T 产品获得FDA 批准用于B 细胞淋巴瘤治疗后,相继已有总计7 款CAR-T产品获批上市,包括美国的5 款和中国的2 款引进产品。除一例靶向B 细胞成熟抗原(BCMA)外,其余几乎均以CD19 为靶点。见表1。
2021 年国内首款CAR-T 产品,奕凯达(阿基仑赛注射液,代号:FKC876)获批用于治疗复发或难治性大B 细胞淋巴瘤成人患者,但是CAR-T 疗法存在局限性,主要用于血液系统肿瘤,对实体瘤的疗效有限。近年来,研究者也在探索CAR-T 免疫疗法用于治疗实体瘤[20]、自身免疫疾病、HIV 感染[21]和心脏病等疾病,展示了广阔的应用空间。
2.3 技术问题及迭代CAR-T 在技术上还存在一些问题,其一是CAR-T 细胞只能杀死含有它们经基因改造后可识别标志物的肿瘤细胞,然而癌细胞停止制造这种标志物产生肿瘤免疫逃逸(Tumor immune escape)的情况并不少见。第二个问题是存在T 细胞耗竭[22],甚至最终被肿瘤细胞抑制。最后,现有的CAR-T 细胞只对它们能轻易到达的血液肿瘤有良好的效果,对于肺部或乳腺中的实体瘤,它们大多无能为力。为了克服这些障碍,斯隆-凯特琳研究所的研究者[23]设计出一种全新的作为一种“微型制药厂”作用的CAR-T 细胞,它可以直接向肿瘤提供有毒的药物载荷,杀死含或不含此种标志物的肿瘤细胞。更重要的是,CAR-T 细胞即使处于耗竭状态时也能产生药物,而且所产生的药物不受癌症抑制。
理论上这些经过基因改造的T 细胞会在体内永久存在,并可以在肿瘤复发时重新发挥作用。但在实践中,许多患者仍然出现肿瘤复发。这是因为肿瘤细胞可以产生更多的雌激素受体结合位点关联抗原9(EBAG9)蛋白,在T 细胞中,EBAG9 蛋白抑制了细胞毒性酶的释放,从而抑制了免疫反应。德国马克斯-德布吕克分子医学中心(MDC)的Armin Rehm 博士和Uta Höpken 博士及其团队[24]在一项发表在JCI Insight 杂志期刊上的研究中发现关闭小鼠体内的EBAG9 基因导致对肿瘤的免疫反应持续增加,这些小鼠还产生了更多的记忆T 细胞。记忆T细胞是免疫记忆的一部分,使人体免疫系统在再次遇到相同的肿瘤抗原后能够做出更好的免疫反应。
在CAR-T 细胞生产中使用病毒也是一个需要应对的问题,因为这种插入、突变,导致产生肿瘤的风险增加。此外,对病毒DNA 的特异性反应往往会阻碍CAR 的表达,而且病毒制备本身经常会产生高额费用。尽管一些不使用病毒的尝试,如使用转座子系统和mRNA 转导,正在用来产生CAR-T细胞,但随机整合和CAR 表达中断导致的CAR-T细胞同质性较低也是一个棘手问题。近期,一些研究表明,通过使用腺相关病毒(Adeno-associated virus,AAV)载体作为模板,可以应用基因组编辑技术来产生基因座特异性整合的CAR-T 细胞。我国科学家于2002 年开发出非病毒的基因特异性靶向CAR-T 细胞[25],并在临床试验中证实它们可安全有效地治疗复发/难治性非霍奇金淋巴瘤,是一项重大进展。
3 CAR-T 应用局限
3.1 患者接受度受限由于CAR-T 的以下几点特性,其在患者中的接受度仍然受限。其一,CAR-T价格过于昂贵,CAR-T 疗法的售价普遍超过30万美元,靶向CD19 的CAR-T 疗法Kymriah 和Yescarta 售价分别为47.5 万美元和37.3 万美元。昂贵的价格一定程度上限制了CAR-T 疗法的普及。由于当前的CAR-T 疗法主要是自体CAR-T疗法,发展通用型CAR-T 疗法,降低售价使尽可能多的人受益,才是这一抗肿瘤技术的最终发展目标。其二,CAR-T 适应证较为有限,目前CAR-T疗法的适应症局限于血液肿瘤,无论是CD19,抑或BCMA,均为血液肿瘤的靶点。其三,CAR-T 治疗风险较高,由于T 细胞在短期内大量被活化,细胞因子的释放在短期内呈爆发式增长,迅速引发免疫反应的同时带来严重的细胞因子释放综合征(Cytokine release syndrome,CRS)。此外,3 级及以上的神经毒性(NT)是另一个重要的风险,临床表现包括幻视、谵妄等。其四,制备周期较长,目前获批上市的5 款CAR-T 疗法均为自体CAR-T 疗法,对于自体CAR-T 疗法而言,平均制备周期须2~6 周。
3.2 实体瘤应用受阻虽然CAR-T 在白血病和淋巴瘤等血液系统肿瘤中取得了令人瞩目的效果,但其仍有许多技术难题仍待解决,包括CRS、NT、脱靶效应等。而且,CAR-T 在实体肿瘤中效果仍不甚理想。实体瘤中较少表达肿瘤特异性抗原(Tumor specific antigen,TSA),高表达的抗原多属于肿瘤相关抗原(Tumor-associated antigen,TAA),其在正常组织中也有表达,这就带来了很高的脱靶风险,使安全性成为关键问题。
现实情况是实体瘤存在高度异质性和复杂的微环境,即便有安全的靶点,能否保证疗效也是一个不小的挑战。
第一,在实体瘤中CAR-T 的转运和浸润受阻。实体瘤具有干扰T 细胞转运进行免疫逃逸的机制。一方面,与血液肿瘤细胞呈分散状不同,实体瘤往往形成坚实的团状物,加上丰富的肿瘤相关成纤维细胞(Cancer-associated fibroblasts,CAFs)和血管,形成了一层天然的物理屏障[26]。另一方面,一些实体瘤会抑制某些趋化因子的分泌。趋化因子与其受体的相互作用会促进T 细胞向肿瘤微环境的迁移。同时,CAR-T 细胞表面也缺乏与实体瘤分泌的趋化因子相匹配的相关受体,造成CAR-T 对肿瘤部位的归巢能力差。第二,肿瘤微环境存在免疫抑制。研究发现,靶向肿瘤微环境(Targeting tumor microenvironment,TME)中低pH、低氧、高渗透,同时存在免疫抑制机制[27],极不利于T 细胞的存活和发挥免疫效力。TME 中存在免疫抑制细胞,如Treg、骨髓源异质性细胞(MDSC)和M2 型巨噬细胞。这些免疫抑制细胞[28]在实体瘤内会释放转化生长因子β(TGFβ)和白细胞介素-10(IL-10)等细胞因子,降低回输后CAR-T 的抗肿瘤效果。第三,内源性T 细胞存在抑制信号。T 细胞的免疫活性存在内源性调节机制,当T 细胞过度活跃时,PD-1 和细胞毒性T 淋巴细胞相关抗原-4(CTLA4)等分子发挥作用,维持免疫平衡。在CAR-T 细胞被抗原激活后,PD-1 和CTLA4 与相关配体相结合,抑制T细胞的增殖和相关细胞因子的分泌[29]。因此,内源性T 细胞抑制信号也会降低CAR-T 的抗肿瘤活性。
4 CAR-T 技术改良进展
4.1 扩大适应证针对适应证有限的瓶颈,目前多家单位相继开展了针对实体瘤CAR-T 药物的研发。处于临床阶段的实体瘤靶点主要包括磷脂酰肌酵蛋白聚糖-3(GPC3)[30]、Claudin 18.2[31]等,这些靶点普遍具有仅在特定细胞表面表达的特点,如Claudin 18.2 仅在人胃上皮短寿细胞表面表达。科济药业的CT041 为一款针对胃及胃食管部位肿瘤的自体靶向Claudin 18.2 的CAR-T 产品,目前处于Ⅰ期临床。考虑到实体瘤肿瘤微环境更为复杂,研究者在预处理上,推出环磷酰胺+氟达拉滨+白蛋白紫杉醇的FNC 方案,有望进一步改变肿瘤微环境,提高药物在实体瘤组织中的渗透性和持久性。
此外,在CAR-T 细胞上增加表达特定抗肿瘤的细胞因子(如IL-12)[32],可吸引NK 细胞和巨噬细胞浸润到实体瘤部位,有望进一步增强CAR-T细胞的有效性。与免疫检查点抑制剂的联用方案是另一个重要的发展方向,可进一步规避实体瘤的免疫逃逸。
4.2 控制治疗毒性CAR-T 细胞治疗显示的毒性和副作用表明,需要制定一些控制程序来调节CARs的活性。大量的方法已经被用来控制CAR-T 细胞的安全性,其中包括通过安装自杀开关快速清除注入的细胞,这种开关可以由小分子或抗体控制。常用的自杀开关[33]包括诱导型caspase-9(iCasp9)、单纯疱疹病毒中的胸苷激酶(HSV-TK)和自杀表位。然而,这样的自杀开关清除了所有的治疗CAR-T 细胞,从而降低了抗肿瘤反应。因此,不清除CAR-T细胞的非细胞毒性可逆系统正在开发中,并具有保持细胞毒性和控制毒性反应之间平衡的潜力。
对此,研究者做出了以下努力:第一,探索使用全人源的CAR 片段[34]代替传统人鼠嵌合片段,提升药物安全性,如科济药业的CT053 凭借全人源CAR 结构,三级及以上CRS 和NT 风险较小;第二,探索在胞内共刺激结构域的选择上,尽量使用4-1BB 而非CD28[35],毕竟早期Juno 的JCAR015 的失败部分源于CD28 共刺激域的使用,T 细胞短期内大量的扩增提升了药物的风险;第三,探索采用创新型的技术如在CAR-T 细胞中引入自毁机制[36],当CAR-T 细胞接触特定配体后发生细胞凋亡,或在CAR-T 细胞表面表达肿瘤抗原及给患者接种单克隆抗体消灭过量的CAR-T 细胞。
4.3 缩短制备周期针对制备周期较长的问题,异体(通用型)CAR-T 细胞是其中的一个解决方案[37]。但对于异体CAR-T 细胞而言,移植物抗宿主病(Graft versus host disease,GVHD)是不得不面对的风险。针对GVHD,可通过CD52 单抗联合化疗清除人体淋巴细胞,或通过CRISPR 技术敲除内源性TCR 和MHC[38],降低GVHD 的风险。此外,许多单位也在探索缩短CAR-T 细胞产品制备周期的方法,如亘喜生物的FasTCAR 平台可将三条主要的制备路径(激活、转导、扩增)合并为一条可同时发生的激活-转导路径,将2~6 周的制备周期缩短至隔天生产。
4.4 开发新靶点实体瘤中常见的TAA 靶点包括CEA、HER2、GPC3、EpCAM 等,TSA 靶点较少,严重限制了CAR-T 在实体瘤中的应用。除了继续寻找和开发针对TSA 靶点的CAR-T 疗法,还可以对CAR-T 细胞进行改造,提高对肿瘤抗原的识别能力。2021 年,WendellLim[39]教授团队在CAR-T 细胞中植入了synNotch 系统,在synNotch 的调控下,识别相关TAA 的CAR 只会在迁移到肿瘤中的T细胞上表达,而不会攻击正常组织中的细胞,大大提高了CAR-T 识别抗原的特异性。Carl H.June 团队和Andy J.Minn 团队植入RNARN7SL1 和外来抗原到肿瘤细胞中,构建多装甲CAR-T,从而提高对肿瘤细胞特异性[40]。此外,除了对CAR-T 细胞本身进行改造,科学家还利用外界条件提高CAR-T对肿瘤细胞的特异性识别。Nature Biomedical Engineering[41]上发表的一篇研究进展显示将超声波与CAR-T 细胞疗法相结合,可以在保护正常组织的同时摧毁肿瘤组织,大大提高了CAR-T 疗法的安全性。
目前针对两个靶点的双特异性CAR-T 已在血液肿瘤领域得到应用,可以提高CAR-T 对抗原的识别能力,增加疗效和安全性,降低肿瘤逃逸的风险。靶向两种或者多种不同TAA 的CAR-T 疗法也是治疗实体瘤的重要方向。比如设计针对多个TAAs 的双特异性CAR-T[42]是可以采用的,利用特异性和广泛靶向性,提高CAR-T 细胞的适应度,包括活化潜能、增殖能力和生存能力,延长CAR-T 细胞在患者体内的生存时间。最近,Nature Cancer 上发表了一篇针对神经母细胞瘤(Neuroblastoma,NB)模型的双靶点CAR-T 研究进展[43],该研究靶向GD2 和B7-H3 两种NB 相关抗原,并提供CD28 和4-1BB 共刺激,在小鼠中实现了快速和持续的抗肿瘤作用,并能防止因抗原密度较低而产生的肿瘤免疫逃逸。
4.5 促进T 细胞浸润治疗血液肿瘤时CAR-T 通常采用静脉回输,在实体瘤中,为改善CAR-T 的浸润和转运难题,可采用瘤内给药的方式。瘤内给药方式在胸膜恶性间皮瘤、头颈癌和恶性胶质瘤的研究中都得到了较好的效果。
近年来针对趋化因子的研究给增强CAR-T 对实体瘤的浸润带来了新希望,趋化因子对于T 细胞浸润发挥着重要作用。2021 年,广州百暨基因研发团队[44]首次研发出经过CXCR5 修饰的CAR-T细胞,其体内示踪实验结果显示可以更好地定向迁移和渗透至肿瘤病灶处,极大减轻了潜在的肿瘤外毒性。此外,也有一些研究靶向肿瘤基质细胞中表达的成纤维细胞活化蛋白(Fibroblast activation protein,FAP),可阻断基质的形成。
TME 中的免疫抑制作用是影响CAR-T 在实体瘤中发挥效力的关键因素之一。TME 产生免疫抑制作用的主要原因是免疫抑制细胞因子、免疫抑制细胞的存在和免疫激活因子的缺乏。研究者通过修饰CAR-T 细胞,使其过度表达促进炎症的细胞因子(如IL-12、IL-15 和IL-18),此为被称为“装甲”CAR-T 细胞(armoured CAR-T cells)的第四代CAR-T 细胞,具有调节局部微环境的作用。研究证实,在卵巢癌中,IL-12[45]可以提高T 细胞的增殖和存活能力,抵抗凋亡和PD-1 诱导的功能抑制。此外,对分泌IL-18 的CAR T 细胞的相关研究表明[46],这些细胞增殖和浸润能力提高,并能募集内源性免疫细胞以调节TME。除了TME 中的免疫抑制作用,T 细胞还存在内源性免疫抑制机制,目前主要使用基因沉默、PD-1 开关受体以及与PD-1 抑制剂联用等手段来避免内源性抑制信号[47]。
4.6 联合传统治疗CAR-T 治疗血液肿瘤,已获较高缓解率,但是患者往往会出现疾病复发的现象。CAR-T 对抗肿瘤的疗效受到T 细胞在体内的激活和扩增、杀伤效果和持久性等多种因素的影响,也与肿瘤类型和患者个体差异息息相关。面对高度复杂的实体瘤,想要通过单一的CAR-T 疗法来平衡这些因素显得举步维艰。除了强化CAR-T 本身对肿瘤细胞的杀伤作用,还应努力探索应对CAR-T扩增和持久性等难题的策略[48]。CAR-T 联合传统的放化疗、免疫检查点抑制剂、疫苗和溶瘤病毒等手段显示出广阔的应用前景,有望通过直接增强T细胞功能、招募内源性免疫细胞和重塑TME 等途径实现治疗实体瘤的效果。
5 CAR 的发展和展望
NK 细胞及巨噬细胞也是重要的免疫细胞类型。随着对其进一步研究发现,与T 细胞类似,他们也可以转导表达CARs。CAR-NK 细胞往往直接采用CAR-T 细胞的设计,由于NK 的细胞毒性是由“缺失自我”的识别引发的,因此NK 细胞尤其具有杀死MHC 下调的肿瘤细胞的能力。有临床前研究提示,以PSCA 为靶点的CAR-NK 细胞针对前列腺肿瘤细胞的裂解率显著提高[49]。
许多实体肿瘤中富含M2 样抑炎性的巨噬细胞,表明巨噬细胞对实体肿瘤具有天然的募集特性。研究者用CAR 修饰巨噬细胞,制成CAR-M,使巨噬细胞获得M1 样促炎性表型,而后不仅能吞噬杀伤肿瘤细胞,还能逆转实体肿瘤的免疫抑制微环境[50]。有研究者利用Ad5f35 型腺病毒载体感染人外周血来源的单核巨噬细胞,构建靶向Her2 的CAR-M,然后静脉转输到乳腺癌和卵巢癌的小鼠模型中。结果显示,CAR-M 不仅可以直接吞噬、杀伤肿瘤细胞,还能逆转免疫抑制微环境,并且还可以递呈肿瘤抗原给T 细胞,增强T 细胞的抗肿瘤能力。
此外,最新研究表明利用CTLA-4 细胞质尾部的内吞特性重新编码CAR,构成单体、双链或三链CTLA-4 胞质尾部(CCTs),可以增强其抗肿瘤功效[51]。
CAR 的发展需要控制成本、合理定价,价格问题是患者最为关注的。多家单位正探索门诊用药的可能性,目前,传奇生物的JNJ-4528 的CARTITUDE 试验[52]纳入了1 名门诊用药的患者,探索门诊用药的可能性,以期降低治疗费用。
许多研究者正在开展对通用型CAR-T 的探索。由于当前的CAR-T 疗法主要采用患者自己的T 细胞,相当于定制药范畴,因此价格高昂,通用型CAR-T 的探索对于其普及非常重要。
另外监管方面要完善监管机制,目前对CAR-T细胞治疗汲取国外经验作为药品进行监管,需完善并进行临床试验审批的流程,推动CAR 的规范化、流程化。相信在不远的将来,CAR 在肿瘤的治疗中将起到越来越重要的作用,必将为人类对抗肿瘤做出重要的贡献。
猜你喜欢
中老年保健(2021年3期)2021-12-03
中国生殖健康(2020年7期)2020-12-10
中国外汇(2019年18期)2019-11-25
哲学评论(2017年1期)2017-07-31
领导决策信息(2017年9期)2017-05-04
领导决策信息(2017年9期)2017-05-04
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年6期)2015-07-01
医学研究杂志(2015年7期)2015-06-22
癌变·畸变·突变(2015年3期)2015-02-27
