
转录组数据分析揭示非洲猪瘟病毒感染后的宿主免疫应答机制及关键效应因子
2023-09-19 00:47:24陈涛苟士学郑伟金银戈蒋美玲罗贤周小青
生物化工 2023年4期
陈涛,苟士学,郑伟,金银戈,蒋美玲,罗贤,周小青*
(1.五邑大学 生物科技与大健康学院,广东江门 529020;2.中国科学院 广州生物医药与健康研究院,广东广州 510530)
非洲猪瘟(African Swine Fever,ASF)是一种高传染性出血性疾病,主要感染对象是家猪和野猪。猪感染ASF 后的临床症状表现不尽相同,从死亡率100%的急性症状到慢性甚至无症状均有[1]。非洲猪瘟病毒(African Swine Fever Virus,ASFV)是一种具有包膜的大型DNA 病毒,是非洲猪瘟病毒科非洲猪瘟病毒属的唯一成员。ASFV 基因组信息量巨大,具有颗粒结构复杂、免疫逃逸手段多样高效、定期重组自身基因组等特点,这些都给疫苗的研制造成了很大困难[2]。迄今为止,人们还没有研发出可用于预防或治疗非洲猪瘟的有效疫苗和抗病毒药物,因此对ASFV 的传染机制和致病机制进行深度探索具有十分重要的意义[3]。本研究利用生物信息学方法,对非洲猪瘟感染样本的转录组数据进行分析,旨在探究与非洲猪瘟感染机制和致病机制相关的关键基因,为ASFV 的疫苗和药物开发提供新的候选靶点。
1 材料与方法
1.1 数据来源
基因表达综合(Gene Expression Omnibus,GEO)公共数据库(https://www.ncbi.nlm.nih.gov/geo)收录了全世界大部分的表达谱数据及高通量测序数据。本研究通过筛选下载了GSE132905(GPL26793)的转录组数据,该数据包含感染ASFV 后3 h、6 h、9 h、12 h、15 h、18 h 的6 个时间点的转录组数据,每个时间点有3 个重复。
1.2 主成分分析
本研究利用主成分分析(Principal Component Analysis,PCA)对各时间样本之间的情况进行分析。PCA 是一种常用的多元数据分析方法,在生物信息学领域中被广泛应用。它可以将高维数据映射到低维空间,并保留数据的主要特征。这种方法可以用于寻找数据的主要变化模式,帮助研究人员更好地理解数据。
1.3 差异基因
针对GSE132905 数据,首先根据GPL26793 平台提供的注释文件,将探针ID 转换为对应的基因ID,并根据数据集中提供的差异倍数值|log FC|>1 作为条件筛选差异基因。
1.4 聚类分析
本研究所选择的数据集中的所有样本都是以不同时间点为实验条件进行测序的,所以可以选择采用基于时间变量的聚类分析模块DEG-Patttern 分析方法进行差异基因的分析,该分析是使用DEGreport包中的degPatterns 函数区分差异基因的不同表达模式。
1.5 功能富集分析
为了描述感兴趣基因列表的分子功能或通路,使用gprofiler2 进行功能富集分析。gprofiler2 是一个生物信息学工具包,用于对基因和蛋白质序列进行功能和富集分析,它可以帮助研究人员在大规模基因表达数据中寻找生物学过程、代谢途径、信号通路等方面的富集。
1.6 PPI 互作网络分析
为了探究关键基因潜在的相互作用,对各时间点的差异基因取交集,并将交集基因上传到STRING 数据库(https://cn.string-db.org/)构建蛋白质互作网络(PPI)的可视化结果。
2 结果与分析
2.1 差异分析
对数据集中的各样本进行主成分分析(PCA),结果表明感染初期的样本较集中,感染后期的样本聚类较集中,反映出实验样本具有可靠性(图1)。根据|log FC|>1 的条件筛选得到918 个差异基因,并以柱状图的形式展示各时间点的差异基因数量,发现差异基因主要集中在感染后期,说明感染的时间越长,样本之间的差异越大,差异基因的数量越多(图2)。

图1 主成分分析图

图2 差异基因柱状图
2.2 聚类和富集分析
对筛选得到的差异基因,使用DEGreport包(https://rdrr.io/github/lpantano/DEGreport/man/degPatterns.html)的degPatterns 函数进行DEG-Pattern 聚类分析,聚类结果显示聚类4 的535 个基因和聚类2 的258 个基因表达分为两个趋势:一种是随着感染时间的增加,基因的表达逐渐下调;另一种是随着时间的增加,基因的表达逐渐上调,说明该部分的基因具有研究意义(图3、图4)。然后利用gprofiler 包[4]中的gost 函数对聚类中的差异基因进行富集分析,富集分析结果发现聚类4 的基因主要与抗病毒反应和免疫反应等通路相关,符合在感染初期由于病毒的入侵迅速激起免疫反应,而随着感染时间的增加,病毒会对免疫系统产生影响从而降低基因的表达。聚类2 的基因主要与免疫细胞的增殖相关,猜测是由于随着感染时间的增加,体内病毒载量增加,生物体会促进免疫细胞的增殖(图5、图6)。
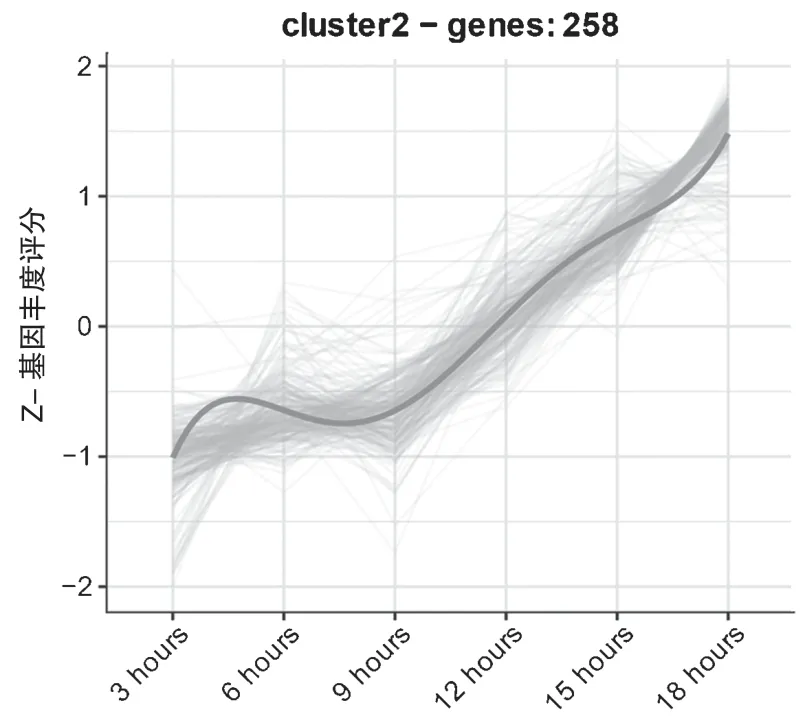
图3 DEG-Pattern 聚类2 结果图
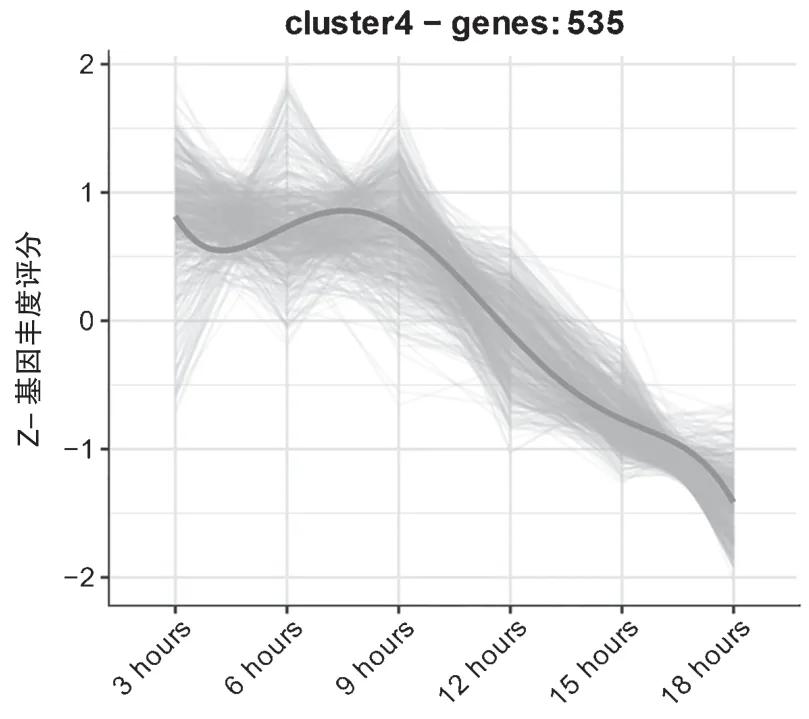
图4 DEG-Pattern 聚类4 结果图

图5 DEG-Pattern 聚类2 富集分析图

图6 DEG-Pattern 聚类4 富集分析图
2.3 关键基因筛选
对各时间点的差异基因取交集,共获得12 个关键基因,表明这些基因参与病毒入侵的全程,对于病毒入侵机制的研究具有一定的意义。用柱状图对这些基因在各时间的表达进行展示,发现基因的表达与聚类的结果一致。UBE2L6、ISG15、ISG20、HERC6基因的表达在感染初期逐渐升高,随后保持不变;APOL3、IL1RN、DOX58、IFIT1、IFIT2、RSAD2、RHPN2、CXCL10 基因的表达在感染的初期高表达,随着感染时间的增加表达逐渐降低,猜测这部分基因可能在感染初期产生反应,基因的表达快速升高,随着感染时间的增加,病毒对免疫系统产生影响,导致基因的表达下调(图7)。

图7 关键基因在各实验组的表达图
2.4 关键基因鉴定
利用STRING 蛋白互作网络在线数据库(https://cn.string-db.org/)分析筛选得到的12 个关键基因潜在的相互作用。蛋白质互作网络图(图8)结果表明,在12 个关键基因中有9 个关键基因存在相互作用,这9个基因分别是IFIT1、IFIT2、ISG15、ISG20、CXCL10、DDX58、HERC6、UBE2L6、IRG6(RSAD2)。查阅NCBI 数据库发现,这9 个基因的表达主要与细胞的抗病毒作用和先天免疫反应及免疫细胞的增殖相关:IFIT1、IFIT2 是编码干扰素诱导的四肽重复蛋白;ISG15、ISG20 是干扰素刺激基因,与免疫系统的信号通路相关;HERC6、UBE2L6 是泛素酶,与免疫蛋白的合成相关;CXCL10 是编码趋化因子,与NK细胞、T 细胞的迁移相关;DDX58 可编码RNA 受体RIG-I,与先天免疫系统和干扰素的产生相关;IRG6(RSAD2)可编码抗病毒蛋白,能够在细胞抗病毒反应和先天免疫中发挥作用。

图8 关键基因蛋白互作网络分析图
3 讨论
综合当前的研究发现,干扰素相关基因在宿主抵抗病毒时发挥着至关重要的作用[3,5]。HEIDEGGER 等[6]揭示了DDX58 编码的RNA 受体RIG-I 蛋白能够诱导促炎性细胞因子和IFN-I 产生;PICHLMAIR 等[7]发现IFIT 家族蛋白能够调节转录起始、细胞增殖与细胞迁移等多种生命活动;BOGUNOVIC 等[8]发现干扰素刺激基因ISG15、ISG20 能够刺激T 淋巴细胞与NK 细胞释放IFN-γ,在抵抗病毒、细菌等先天免疫和适应性免疫中发挥重要作用;FREITAS 等[9]发现病毒在复制的过程需要泛素结合酶E2 等多种酶的参与;GAO 等[10]证实UBE2L6 可能被干扰素上调,从而产生抗病毒作用。
本研究对公用数据库的数据进行挖掘,首先对数据进行主成分分析,发现感染后不同时间的样本聚类明显,反映了转录组数据的可靠性。使用差异分析和DEG-Pattern 聚类分析相结合,共筛选出918 差异基因,对这些差异基因进行聚类分析,发现第一部分差异基因在刚感染时表达量增加,随着感染时间的增加表达量呈现逐渐减少的趋势,第二部分差异基因在感染后表达量呈现逐渐增加的趋势。对这两部分的差异基因进行富集分析,结果表明第一部分的差异基因主要与细胞的抗病毒反应及免疫反应等通路有关,第二部分的差异基因主要与免疫细胞的增殖通路有关。对不同时间点的差异基因取交集,得到了12 个关键基因。将这些基因上传到STRING 蛋白互作网络在线数据库进行分析,蛋白互作网络分析结果显示有9个基因存在相互作用,并通过检索NCBI 数据库发现这9 个基因大多数都参与干扰素的生成、泛素酶的合成、先天免疫反应、抗病毒机制、病毒的复制、免疫细胞的增殖等。这些基因的异常表达很可能是由于非洲猪瘟病毒的入侵对其产生了干预。笔者筛选出来的多个表达异常的基因,如DDX58、IFIT1、IFIT2、ISG15、ISG20 和UBE2L6 等,已经在非洲猪瘟相关文献有过类似报道[9],说明这些基因是研究非洲猪瘟感染机制和致病机制的潜在靶点基因。
综上所述,通过生物信息学分析和蛋白互作数据库筛选,可鉴定出有助于揭示ASFV 潜在感染机制和致病机制的关键基因,为ASFV 的疫苗和药物开发提供新的候选靶点。虽然本研究得到的关键基因需要进一步通过实验进行验证,但基于数据库进行的生物信息学的数据挖掘还是提供了可靠的方法,缩小了对于关键基因的筛选范围,节省了大量的资源成本和人力成本。
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
中学生数理化(高中版.高考理化)(2021年6期)2021-07-28 06:21:04
心电与循环(2020年1期)2020-02-27 07:48:24
幸福(2019年12期)2019-05-16 02:27:44
猪业科学(2018年8期)2018-09-28 01:27:52
兽医导刊(2016年6期)2016-05-17 03:50:36
湖北农业科学(2014年3期)2014-07-21 10:25:00
NBA特刊(2014年7期)2014-04-29 00:44:03
当代畜禽养殖业(2014年3期)2014-02-27 07:58:45
中国商人(2013年1期)2013-12-04 08:52:52
