
pH 区带精制逆流色谱法分离制备广西地不容非药用部位中生物碱
2023-09-19 02:44张汝胜王亚凤阳丙媛何瑞杰黄永林
中成药 2023年9期
张汝胜,李 霞,王亚凤,阳丙媛,何瑞杰*,黄永林*
(1.桂林理工大学化学与生物工程学院,广西 桂林 541006;2.广西植物功能物质研究与利用重点实验室,广西壮族自治区中国科学院广西植物研究所,广西 桂林 541006)
广西地不容StephaniakwangsiensisLo.是广西特有植物,属于防己科千金藤属多年生草质落叶藤本植物,主要分布于当地西北、西南部,生于石灰岩地区的山地灌丛[1],具有镇痛、抗炎[2]、抑菌[3]、抗病毒[4]、杀虫[5]、治疗阿尔茨海默病[6]等作用。该植物药用部位为根块,是生产颅痛定的原材料,而地上部分也富含生物碱,其中的青藤碱、氧化青藤碱含量很高。为了提高广西地不容整体利用率,创建一种快速高效分离制备其非药用部位中生物碱的方法尤为必要。
高速逆流色谱是一种新型高效的液-液分配色谱技术,主要通过在2 个互不相溶的上下相中的分配比的差异来实现分离,具有无柱污染、无不可逆吸附、进样量大、同一根逆流色谱柱既可用于分析又可用于制备等优点[7]。pH 区带精制逆流色谱法是在普通制备高速逆流色谱的基础上发展起来的特殊逆流色谱分离制备方法[8],特别适用于生物碱、有机酸的制备性分离[9],本实验采用该方法对广西地不容非药用部位总生物碱进行分离制备,共得到2 个高纯度化合物,以期为相关大规模生产提供参考。
1 材料
TBE-300C 型pH 区带精制逆流色谱仪(上海同田生物技术股份有限公司);LC-20AT 型高效液相色谱仪(日本岛津公司);SP-MAX3500FL 型荧光酶标仪(上海闪谱生物科技有限公司)。
阿卡波糖、曲酸、α-葡萄糖苷酶均购自上海源叶生物科技有限公司;酪氨酸酶购自上海麦克林生化科技有限公司。无水碳酸钠、二氯甲烷、正丁醇、甲醇均为分析纯,购于西陇化工股份有限公司。
广西地不容非药用部位于2019 年8 月采自广西桂林恭城瑶族自治县,经广西植物研究所黄俞淞副研究员鉴定为正品,标本(编号20190824) 保存于广西植物功能物质研究与利用重点实验室。
2 方法
2.1 样品溶液制备 取干燥药材500 g,粉碎后用95%乙醇回流提取3 次,每次2 h,合并提取液,减压浓缩至浸膏状态,溶于盐酸(pH=3) 中,过滤得清液,氨水调节pH值至10 左右,二氯甲烷萃取,减压浓缩得到总生物碱8 g,取1.60 g 至试管中,5 mL 三乙胺固定相溶解,即得。
2.2 溶剂体系制备 前期报道,与通过HPLC 法测定K值来确定溶剂系统比较,TLC 法分析Rf值更方便快捷[10-11],本实验采用该方法确定二氯甲烷-甲醇-正丁醇-水(10 ∶6 ∶0.1 ∶4) 作为溶剂体系。
2.2.1 正相置换分离模式 量取二氯甲烷850 mL、甲醇510 mL、正丁醇8.5 mL、水340 mL,置于分液漏斗中,室温下充分混合,静置分层,将上下相分别收集,上相加入10 mmol/L 三乙胺作固定相,下相加入10 mmol/L 盐酸为流动相,超声脱气20 min,即得。
2.2.2 反相置换分离模式 量取二氯甲烷900 mL、甲醇540 mL、正丁醇9 mL、水360 mL,置于分液漏斗中,室温下充分混合,静置分层,上下相分别收集,上相加入5 mmol/L 盐酸作为流动相,下相加入10 mmol/L 三乙胺作为固定相,超声脱气20 min,即得。
2.3 分离与鉴定
2.3.1 分离原理 pH 区带逆流色谱是利用不同化合物之间解离常数、疏水性的差异来实现分离[12],待分离物流出以区带pH 为顺序,而后者与前者pKa值、疏水性相关。以分离生物碱(图1) 为例,疏水性的该成分从洗脱酸中获得质子生成盐而溶于流动相中(位置④),流动相中盐与高pH 值固定相界面接触(位置①),盐被保留碱夺去质子后生成疏水性的生物碱而溶于固定相中(位置②),此时保留碱边界向前推移,生物碱又与洗脱酸界面接触(位置③),疏水性的生物碱重新获得质子生成盐又溶入到流动相中(位置④),不断地重复此循环过程,直到保留碱流出色谱柱为止[13-15]。

图1 pH 区带精制逆流色谱分离原理图
2.3.2 分离过程
2.3.2.1 反相置换模式 将下相以30 mL/min 体积流量泵入高速逆流主机中作为固定相,整个分离管被固定相充满后开启主机,缓慢调节主机转速达到850 r/min 并稳定后,将样品溶液注入进样环,以1.5 r/min 转速泵入流动相,开启紫外检测器、记录仪,在254 nm 波长处收集馏分,TLC合并并低温干燥,得到40~90 min 馏分、化合物2,色谱图见图2。

图2 N-氧化青藤碱pH 区带精制逆流色谱图
2.3.2.2 正相置换模式 HPLC 分析显示,40~90 min 馏分为纯度较低的化合物,故调整洗脱酸浓度(加入10 mmol/L 盐酸),将上相设为固定相,下相设为流动相,从中分离得到化合物1,色谱图见图3。
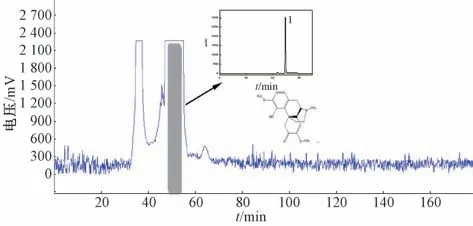
图3 青藤碱pH 区带精制逆流色谱图
2.3.3 HPLC 分析条件 为了得到较好的峰形,实现基线分离,选择甲醇-0.1% 三乙胺体系作为流动相。分析采用Eclipse XDB-C18色谱柱(4.6 mm×250 mm,5 μm);体积流量1 mL/min;柱温40 ℃;检测波长254 nm。色谱图见图4~6。

图4 总生物碱HPLC 色谱图

图5 青藤碱HPLC 色谱图
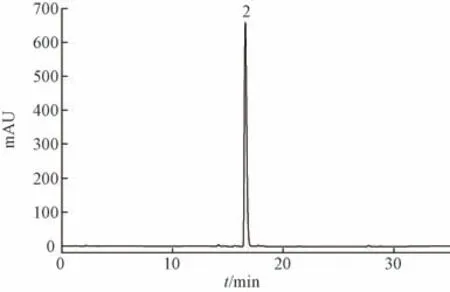
图6 N-氧化青藤碱HPLC 色谱图
2.3.4 结构鉴定 化合物1:白色粉末,HR-ESI-MSm/z:330.170 0 [M+H]+(计算值C19H23NO4)。1H-NMR (500 MHz,CDCl3)δ:6.52 (1H,d,J=8.3 Hz,H-1),6.62(1H,d,J=8.2 Hz,H-2),4.34 (1H,d,J=15.6 Hz,H-5),2.43 (1H,d,J=15.7 Hz,H-5),5.46 (1H,s,H-8),3.16 (1H,brs,H-9),3.01 (1H,brs,H-10),2.69 (1H,dd,J=18.5,5.3 Hz,H-10),2.98 (1H,brs,H-14),1.89(1H,m,H-15),1.84 (1H,m,H-15),2.52 (1H,ddd,J=11.9,4.5,2.1 Hz,H-16),2.06 (1H,td,J=12.0,3.7 Hz,H-16),3.79 (3H,s,3-OCH3),3.48 (3H,s,7-OCH3),2.42 (3H,s,NCH3);13C-NMR (125 MHz,CDCl3)δ:118.3 (C-1),109.1 (C-2),145.1 (C-3),144.8(C-4),49.4 (C-5),194.1 (C-6),152.5 (C-7),115.2 (C-8),56.8 (C-9),24.4 (C-10),130.5 (C-11),122.7 (C-12),40.6 (C-13),46.1 (C-14),36.2 (C-15),47.3 (C-16),56.2 (3-OCH3),54.9 (7-OCH3),42.9 (NCH3)。以上数据与文献[16] 报道一致,故鉴定为青藤碱。
化合物2:白色针晶,HR-ESI-MSm/z:344.150 3 [MH]-(计算值C19H23NO5)。1H-NMR (500 MHz,DMSO-d6)δ:6.57 (1H,d,J=8.4 Hz,H-1),6.82 (1H,d,J=8.4 Hz,H-2),2.62 (1H,d,J=15.5 Hz,H-5),4.17 (1H,d,J=15.4 Hz,H-5),5.72 (1H,d,J=2.2 Hz,H-8),3.99(1H,t,J=4.4 Hz,H-9),3.16 (1H,brs,H-10),3.37(1H,brs,H-10),3.86 (1H,brs,H-14),2.32 (1H,brd,J=4 Hz,H-15),1.77 (1H,brd,J=13.3 Hz,H-15),3.18(1H,brd,J=11.6 Hz,H-16),2.90 (1H,m,H-16),3.72(3H,s,3-OCH3),3.63 (3H,s,7-OCH3),3.35 (3H,s,NCH3);13C-NMR (125 MHz,DMSO-d6)δ:118.5 (C-1),110.7 (C-2),146.2 (C-3),145.2 (C-4),47.7 (C-5),192.0 (C-6),151.9 (C-7),113.7 (C-8),71.5 (C-9),27.4 (C-10),125.8 (C-11),121.2 (C-12),38.7 (C-13),38.4 (C-14),30.8 (C-15),59.8 (C-16),55.9 (3-OCH3),54.6 (7-OCH3),56.2 (NCH3)。以上数据与文献[6] 报道一致,故鉴定为N-氧化青藤碱。
2.4 酶抑制活性研究
2.4.1 酪氨酸酶 在96 孔板中依次加入样品溶液20 μL、酶液10 μL,混匀,37 ℃水浴10 min,加入40 μL 左旋多巴溶液,37 ℃水浴再反应5 min,平行3 次,在475 nm 波长处测定吸光度,计算抑制率,公式为抑制率={1-[(A1-A2)/(B1-B2)] } ×100%,其中A1、A2、B1、B2分别为样品组、背景组、阴性对照组、空白对照组吸光度。
2.4.2 α-葡萄糖苷酶 在96 孔板中依次加入PBS 缓冲液50 μL、样品溶液20 μL、酶液10 μL,37 ℃水浴5 min 后加入20 μL PNPG,继续水浴25 min 后取出,最后加入50 μL Na2CO3溶液终止反应,平行3 次,在405 nm 波长处测定吸光度,计算抑制率,公式为抑制率={1-[(A1-A2)/(B1-B2)] } ×100%,其中A1、A2、B1、B2分别为样品组、背景组、阴性对照组、空白对照组吸光度。
2.4.3 结果分析 图7 显示,青藤碱、N-氧化青藤碱酶抑制活性均存在量效关系,其中前者随着其质量浓度增加对酪氨酸酶的抑制活性大于后者,而后者随着其质量浓度增加对α-葡萄糖苷酶的抑制活性大于前者。表1 显示,与阳性对照组(前者为曲酸,后者为阿卡波糖) 比较,青藤碱对α-葡萄糖苷酶的抑制活性升高(P<0.05),而后者对酪氨酸酶的抑制活性升高(P<0.01),但IC50值均较低。
表1 各化合物对酪氨酸酶、α-葡萄糖苷酶的IC50值(±s)

表1 各化合物对酪氨酸酶、α-葡萄糖苷酶的IC50值(±s)
注:与阳性对照组 (曲酸、阿卡波糖) 比较,* P <0.05,**P<0.01。
样品IC50/(μmol·L-1)酪氨酸酶α-葡萄糖苷酶青藤碱6 330±160.96 400±172*N-氧化青藤碱6 630±178.9**39 000±900曲酸69.6±0.088—阿卡波糖—0.001 24±0.000 015
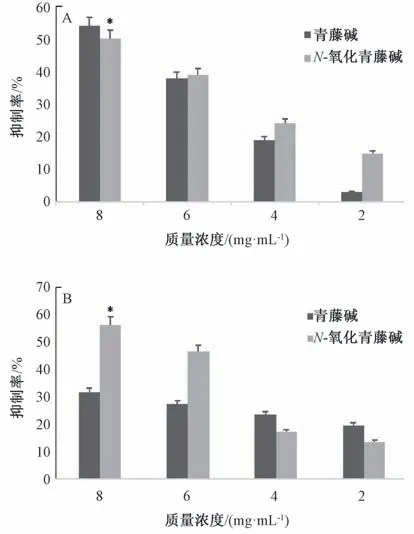
图7 各化合物对酪氨酸酶(A)、α-葡萄糖苷酶(B) 的抑制作用
3 讨论与结论
生物碱是成药性较高的一类化合物,但其分离一直都是天然产物化学研究领域的一大难题。pH 区带精制逆流色谱法主要根据不同化合物在溶剂中的解离常数和疏水性的不同而实现分离,可有效解决普通的柱色谱法存在的吸附严重、拖尾、收率低的问题。本实验采用该方法快速高效地从广西地不容非药用部位中分离到青藤碱、N-氧化青藤碱,可为这2 种生物碱的大规模生产提供指导,也为其他生物碱的分离制备提供新方法。另外,广西地不容非药用部位资源丰富,生物碱含量高(尤其是青藤碱、N-氧化青藤碱),开发潜力巨大,能否成为替代资源仍需进一步研究。
猜你喜欢
基层中医药(2021年11期)2021-03-26
石油工业技术监督(2019年7期)2019-07-30
小学生作文(低年级适用)(2019年4期)2019-04-29
特种油气藏(2018年6期)2019-01-11
天然产物研究与开发(2018年8期)2018-09-10
中成药(2018年7期)2018-08-04
散文诗(2017年18期)2018-01-31
学生天地(2017年10期)2017-05-17
天然产物研究与开发(2014年8期)2014-04-27
长江大学学报(自科版)(2014年2期)2014-03-20
