
一个玉米ZmMs7复等位基因突变体的遗传分析与分子鉴定
2023-09-14 09:33:20曹枭雄刘伊凡周玉强吴宇锦王红武刘小刚黄长玲刘志芳郭晋杰胡小娇
作物学报 2023年11期
曹枭雄 刘伊凡 周玉强 王 婧 吴宇锦 王红武 李 坤 刘小刚 黄长玲 刘志芳 郭晋杰 胡小娇,*
一个玉米复等位基因突变体的遗传分析与分子鉴定
曹枭雄1,2刘伊凡1,2周玉强2王 婧2吴宇锦2王红武2李 坤2刘小刚2黄长玲2刘志芳2郭晋杰1,*胡小娇2,*
1河北农业大学农学院 / 国家玉米改良中心河北分中心 / 华北作物改良与调控国家重点实验室, 河北保定 071001;2中国农业科学院作物科学研究所 / 作物分子育种国家工程研究中心, 北京 100081
我们在自然群体中发现了一个玉米雄性不育突变体(), 命名为。该突变体雄花育性彻底丧失, 花药干瘪皱缩, 没有花粉形成。细胞学分析发现, 与野生型相比,突变体花药在S11期表现出明显的药室收缩, 绒毡层细胞肿胀, 小孢子破裂的表型, 表明突变体绒毡层细胞程序性死亡出现异常, 且花粉败育。遗传分析表明该不育性状受单个隐性核基因控制。为克隆目标基因, 以为母本分别与不同自交系杂交构建F2定位群体, 利用靶向测序技术(GBTS)分析群体基因型, 将基因定位于7号染色体124.95~128.47 Mb之间, 进一步精细定位将该区间缩小到0.68 Mb。生物信息学分析发现, 该区间存在一个已知基因。基因编码PHD-finger转录因子, 在绒毡层发育和花粉外壁的形成过程中发挥重要作用。等位测验分析发现为基因的等位突变体。基因测序结果表明突变体在外显子区存在多处序列变异, 与所报道的基因已知突变体和的突变方式不同, 证明是一个新的基因等位突变体。突变体的发现与鉴定为探讨玉米核雄性不育的分子机制以及育种应用提供了新的材料。
玉米; 雄性不育;; 基因定位; 等位突变
玉米是全球第一大粮食作物, 也是杂种优势利用最成功的作物。杂交种在生长势、生活力、抗逆性、产量和品质上显著优于2个亲本, 因此广泛应用于玉米生产[1]。目前, 玉米杂交制种主要利用人工或机械去除母本雄穗不但费时费力成本高, 且不利于植物生长, 从而降低杂交种的产量[2]。利用玉米雄性不育系生产玉米杂交种可以有效解决以上问题。雄性不育可分为细胞质雄性不育(CMS)和细胞核雄性不育(GMS)[3], CMS又称为核质互作雄性不育, 以CMS为基础的三系制种已成功应用于多种作物的杂交制种, 但是CMS材料存在表型不稳定、易受病原小种专化感染、恢复系少等缺点[3-4], 限制了CMS材料的应用。GMS育性由核基因控制, 可以克服CMS的缺陷, 具有巨大的育种应用潜力。虽然GMS不育系存在保持和繁种困难的问题, 但是随着现代生物技术的快速发展, 转基因和基因编辑技术的广泛应用, 有望将隐性核不育基因有效利用起来[5]。
在拟南芥和水稻中, 已有大量研究解析了调控花药和花粉发育的过程[6-7], 同时也揭示了14个花药的发育阶段。与水稻花药发育过程相比, 玉米花药发育的变化与其高度一致, 同样也可划分为14个时期。花药的发育从雄蕊原基开始, 经过一系列分裂分化, 花药从最外层到最内层形成表皮层、内皮层、中间层和绒毡层4个典型层。绒毡层是4层壁细胞中代谢最为活跃的一层, 在植物体小孢子发育过程中有非常关键的作用[8]。在小孢子发育的前期, 绒毡层细胞会给花粉母细胞提供大分子物质及营养供给以促进其发育[9-10]; 在小孢子发育中期会分泌花粉外壁前体, 如孢粉素是构成花粉壁和花药表皮角质层的重要物质[11]; 在小孢子发育后期绒毡层会分泌花粉外壳或含油层[12-14]。而绒毡层细胞程序性死亡(PCD)也会为小孢子的发育提供蛋白质、脂类等营养物质, 对于花粉的形成、成熟以及育性都有影响[15-17]。在已报道的众多植物雄性不育突变体中, 绒毡层发育缺陷型雄性不育是最多的一类[18]。且在不同植物中, 也有多个与绒毡层发育相关的基因被克隆, 如在拟南芥中发现基因编码一个富含亮氨酸重复序列的受体蛋白激酶(LRR-RPK), 其表达与小孢子母细胞和绒毡层细胞的分化有关, 突变后导致败育[19]; 在水稻中发现bHLH家族转录因子启动其同源基因和的表达。与相互作用来调控绒毡层分化, 促进绒毡层PCD, 然后与相互作用调节绒毡层细胞死亡,突变导致水稻雄性不育[20]; 在玉米中发现基因编码一个bHLH蛋白,由于绒毡层前体细胞不能分化, 导致花粉母细胞发育失败, 最终影响其育性[21]等。
本研究对一个新玉米雄性不育突变体进行了表型鉴定、细胞学观察、遗传分析和基因定位, 最终发现是已知基因的新等位突变体。研究结果为深入解析玉米雄性不育的分子机制奠定了基础, 为细胞核雄性不育的育种应用提供了新材料。
1 材料与方法
1.1 试验材料
玉米雄性不育突变体是自交系也铁21背景的自然突变体。由于雄性不育突变体不能够自交繁殖, 因此我们通过对杂合体植株进行连续自交和表型鉴定来分离和保存突变体。本研究中用于表型鉴定和细胞学分析的材料为杂合植株连续自交5代后分离出的野生型(WT)和突变体。基因定位群体为突变体与自交系B73、Mo17、昌7-2 (C7-2)杂交并自交获得的F2分离群体。用于等位测验的突变体为北京科技大学化学与生物工程学院万向元教授馈赠。
1.2 突变体的表型鉴定与细胞学观察
在玉米抽雄散粉期开展表型鉴定, 对WT和的雄穗、花药和花粉进行比较, 同时调查株高、穗位高、雄穗分枝数、叶长和叶宽等重要农艺性状。细胞学观察以WT及在S8a、S8b、S9、S10及S11五个时期[2]的花药为试验材料, 用FAA固定液保存, 固定后的组织材料进行脱水、透明、浸蜡与包埋, 将包埋好的蜡块切片黏附于载玻片上并干燥, 然后染色并用树脂封片, 自然晾干。再用OLYMPLUS BX53正置显微镜(奥林巴斯, 日本)观察并照相。
1.3 突变体的遗传分析和基因定位
对于突变体表型的遗传分析, 在玉米散粉期统计不同遗传背景的F2群体中可育单株与不育单株的分离比, 并利用卡方测验确定遗传模式。在与B73构建的F2定位群体中随机选取69株不育单株, 利用CTAB法提取群体及双亲的基因组DNA。利用10K GBTS靶向测序技术分析样品基因型[22]。基因初定位采用SNP-index算法, 通过计算每个多态性SNP标记的SNP-index, 即突变体基因型占所有F2样本基因型的频率, SNP-index越接近1, 表明标记与目标基因连锁越紧密[23]。为精细定位目标基因, 我们扩大F2定位群体数量, 并开发5对插入缺失标记(insertion-deletion, InDel), InD124、InD125、InD127、InD128、InD130 (表1), 进一步缩小定位区间。
1.4 基因组DNA提取、PCR扩增和序列分析
用CTAB法提取亲本及突变体的基因组DNA。利用Primer 5软件设计引物(表1), 扩增基因组序列。以WT与基因组DNA为模板, 分别扩增基因片段, 将PCR产物送生工生物工程(上海)股份有限公司测序。使用DNAMAN软件比对分析测序结果, 确定基因突变情况。试验采用南京诺唯赞生物科技有限公司的PCR反应试剂, 扩增体系为2× PCR Mix 10 mL, 正向和反向引物(10 μmol L–1) 0.5 μL, 模板DNA 1 μL, 补超纯水至20 μL。PCR扩增条件为94℃预变性3 min; 94℃变性15 s, 58℃退火15 s, 72℃延伸2 min, 34个循环; 72℃延伸5 min。用1%琼脂糖凝胶电泳检测目的条带。
1.5 RNA提取及基因组织表达分析
基因的组织表达分析以自交系C7-2苗期的叶和根, 拔节期的叶、根和第3节茎间, 抽雄期的叶、根、雄穗、叶耳、雌穗、第3节茎间、花丝及花药为研究材料。用植物总RNA提取试剂盒(天根生化科技(北京)有限公司, Cat# DP432)提取不同组织的RNA。利用反转录试剂盒(天根生化科技(北京)有限公司, Cat# KR118-02)反转录为cDNA, 以cDNA为模板, 设计特异引物20q-F/R, 并以Tubulin ()为内参基因(表1), 在Bio-Rad CFX96实时定量PCR仪上进行荧光定量分析。按2–ΔCT方法[24]计算基因在各个组织中的表达水平, 分析其组织表达模式。

表1 本研究采用的PCR引物
2 结果与分析
2.1 突变体ms20s1的表型鉴定和细胞学观察
在玉米抽雄散粉期, 野生型和突变体植株都能够正常抽雄。野生型雄花花药饱满且颜色较黄, 能够正常开裂及散粉, 自交可正常结实; 而不育株花药干瘪较小且颜色较淡, 颖壳不开裂, 花药不外露(图1-C, D)。作母本与其他野生型植株杂交后可正常结实。对野生型和突变体的花药进行I2-KI染色, 发现野生型花粉粒呈黑褐色, 而花药中没有花粉粒, 表明突变体为无花粉型雄性不育(图1-E, F)。进一步对比野生型与突变体植株, 发现在株高、雄穗主轴长度和雄穗分枝数上有显著差异外(表2), 其他农艺性状和野生型植株无明显差异(图1-A, B)。
为了确定花药发育异常产生的时期, 我们对玉米不同发育时期的花药进行了石蜡切片和细胞学观察。花药横切面观察表明, 在S8a~S8b时期, WT和花药中小孢子母细胞开始减数分裂, 依次产生二分体和四分体, 绒毡层细胞质浓缩, 着色加深, 表型没有明显差异; S9时产生的小孢子被释放出来, WT和花药发育开始出现差异; S10时小孢子开始液泡化, WT相较于来说更为饱满,液泡收缩更早。S11时WT花药小孢子囊泡开始逐步消失, 绒毡层细胞开始退化, 而药室开始收缩, 绒毡层细胞降解异常, 小孢子破裂退化(图2)。
A: 散粉期野生型(WT)与突变体植株的形态比较, 标尺为20 cm; B: 散粉期野生型(WT)与突变体的雄穗比较, 标尺为20 cm; C、D: 野生型(C)与突变体植株(D)的花药比较, 标尺为1 mm; E、F: 野生型(E)与突变体(F)被I2-KI染色后的花粉粒, 标尺为200 μm。
A: the comparison of plant morphology between wild type andmutant at anthesis stage, bar: 20 cm; B: the comparison on tassel traits between the WT andmutant at anthesis stage, bar: 20 cm; C, D: the comparison of the anther morphology between the WT (C) andmutant (D), bar: 1 mm; E, F: pollen grains of WT (E) andmutant (F) stained with I2-KI, bar: 200 μm.
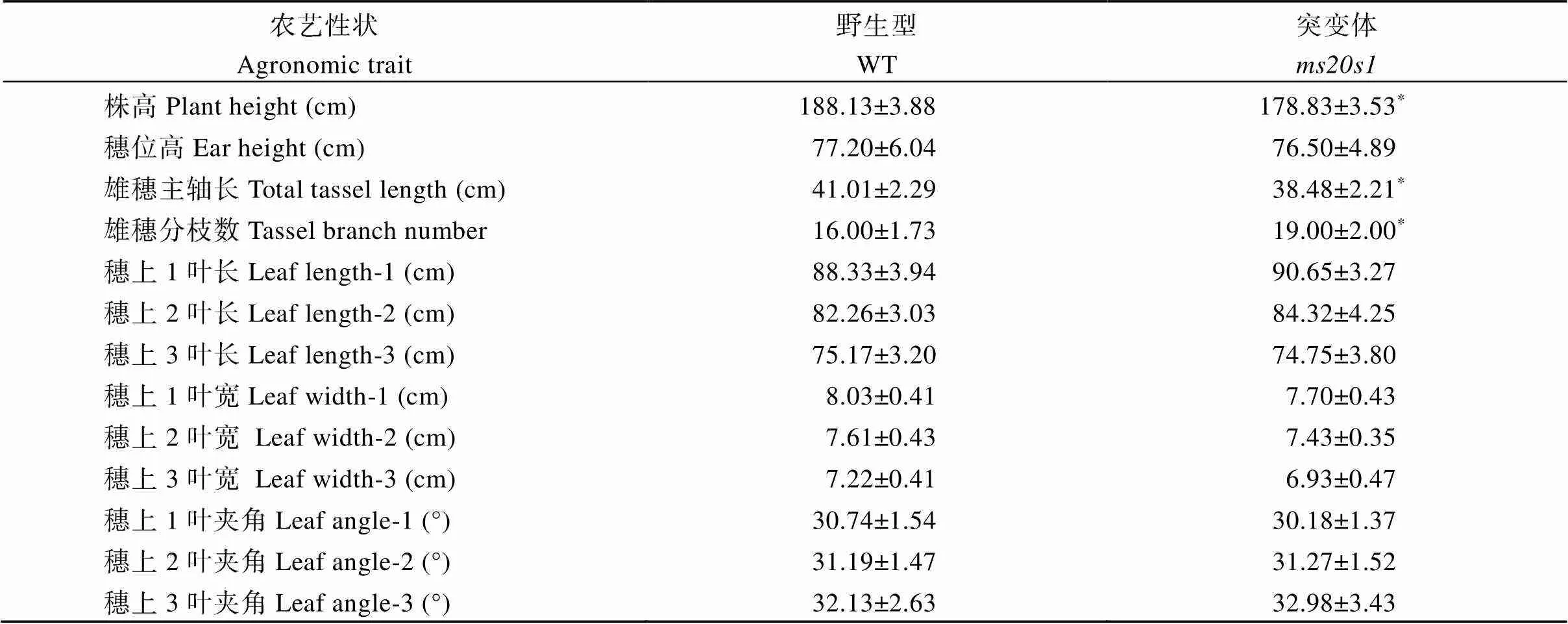
表2 野生型和突变体重要农艺性状比较
*表示在< 0.05水平差异显著。*means significant difference at< 0.05.
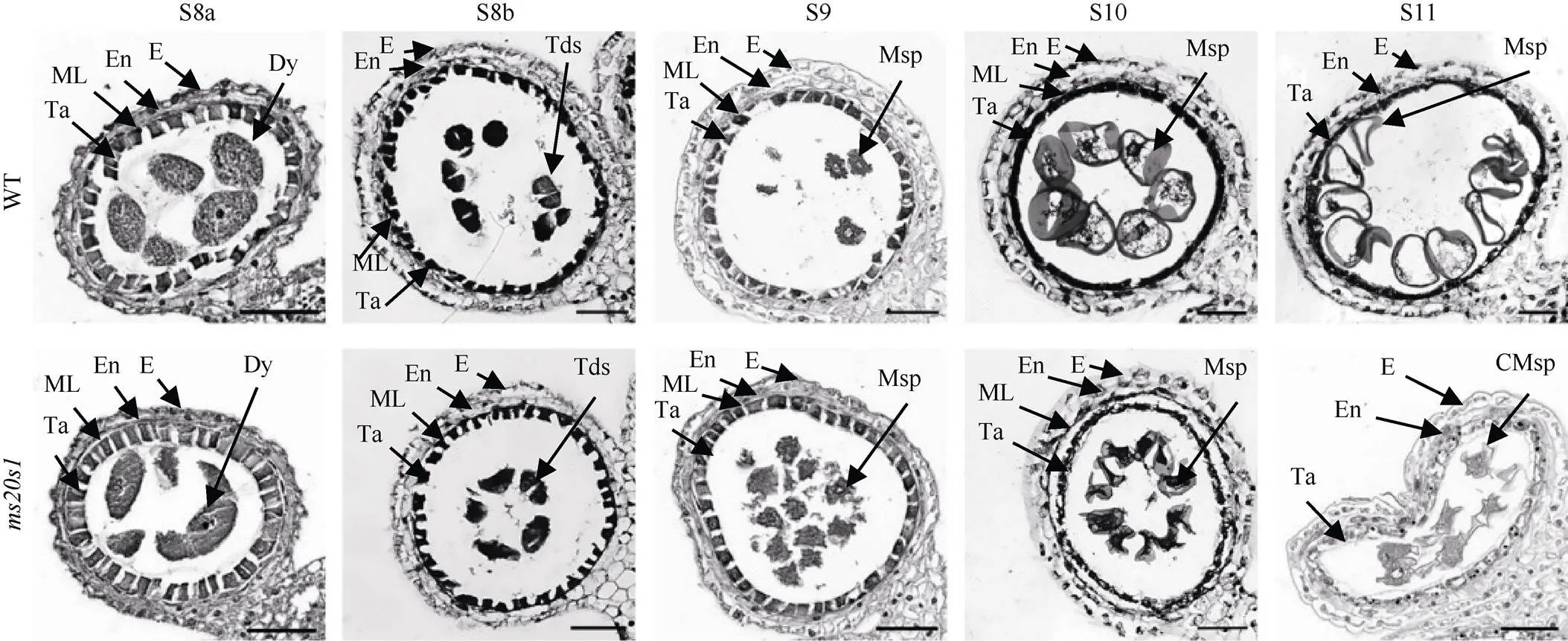
图2 WT和ms20s1突变体玉米花药(S8a~S11期)的横切面分析
E: 表皮层; En: 内皮层; ML: 中间层; Ta: 绒毡层; Msp: 小孢子; CMsp: 收缩的小孢子; Dy: 二分体; Tds: 四分体。标尺为50 μm。
E: epidermis; En: endothecium; ML: middle layer; Ta: tapetum; Msp: microspore; CMsp: collapsed microspore; Dy: dyad; Tds: tetrads. Bar: 50 μm.
2.2 ms20s1的遗传分析与基因定位
将该突变体分别与B73、Mo17、C7-2进行杂交, F1均表现为野生型。统计F1自交得到的3个F2群体中野生型和突变体的数目, 经卡方检验其分离比都符合3∶1 (表3), 表明雄性不育性状是受单个隐性核基因控制。为克隆该基因, 以B73为遗传背景的F2分离群体为材料, 利用CTAB法提取69株不育植株的叶片及其亲本DNA, 利用10K GBTS靶向测序技术分析样本基因型。去除杂合及双亲之间的无多态性位点, 数据分析表明亲本间存在多态性的SNP标记数量为4745个, 占总位点数的47.45%, 平均每条染色体上的数目为474个, 其中1号染色体最多为850个, 6号染色体最少为361个(图3)。分析所有多态性SNP位点对应的SNP-index在全基因组水平上的变化, 发现在7号染色体上124.95~128.47 Mb区间可能与目标性状连锁(图3), 增加F2群体个数到192, 同时开发5个InDel标记(InD124、InD125、InD127、InD128、InD130), 最终将基因定位在7号染色体的InD127和InD128标记之间, 左侧标记处交换单株数为4, 右侧标记处交换单株数为3, 物理距离为0.68 Mb (图4-A)。利用MaizeGDB (https://www.maizegdb.org/)网站检索候选区段的基因及其功能注释, 发现该区间内共有13个蛋白编码基因, 其中包含一个已经报道的玉米雄性不育基因()。
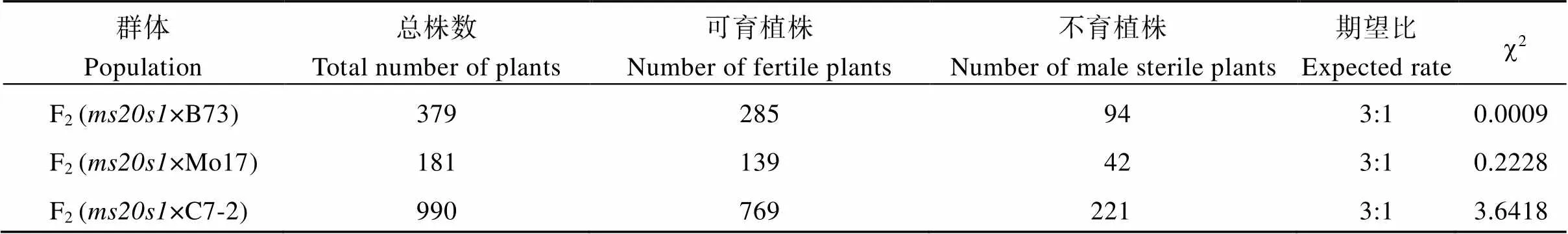
表3 F2群体表型分离的卡方检测
c2(0.05) (1)= 3.84.
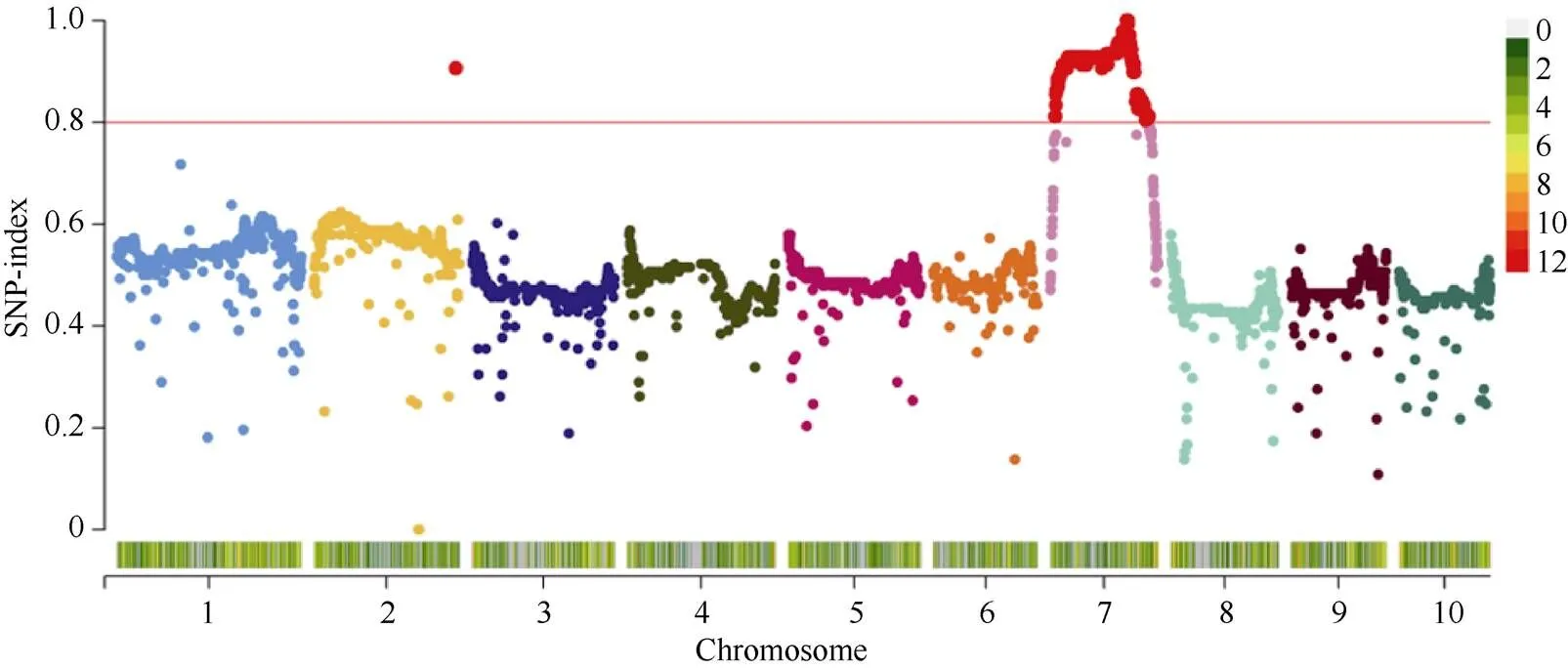
图3 SNP-index全基因组频率分布图
2.3 ms20s1与ms7-6007的等位测验
为了验证是否为基因的等位突变, 我们利用基因的已知突变体杂合基因型单株做父本与不育单株杂交, 同时再以杂合基因型单株作父本与不育单株作杂交, 在这2个杂交后代群体中均出现了雄性不育植株(图5-A), 观察其花药及被I2-KI染色后的花粉, 证实其不育表型(图5-B, C)。分别统计2种杂交方式后代不育单株和可育单株数量。以为母本的等位测验中, 共调查了196株, 表现为野生型表型的有96株, 表现为突变体表型的有100株。以为母本的等位测验中, 共调查了296株, 表现为野生型表型的有151株, 突变体表型的有145株。经卡方检验分析表明野生型与突变体植株的分离比均符合1∶1, 证明是新的等位突变体(表4)。
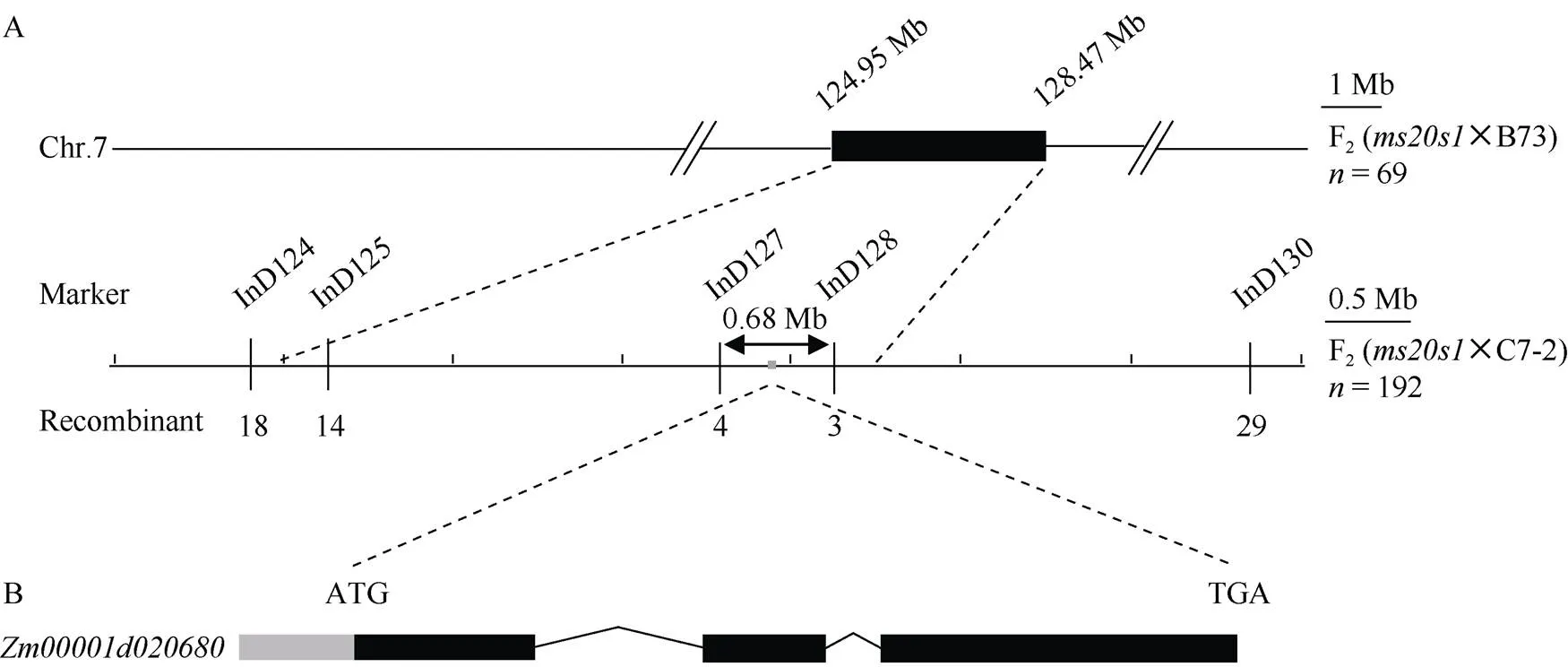
图4 ms20s1的精细定位和ZmMs7基因的结构示意图
A:的精细定位。Marker: 标记名称; N: 群体大小; Recombinant: 重组单株数; B:基因的结构示意图。灰色框代表5¢非翻译区; 黑色框代表外显子区, 横线代表内含子区。
A: fine mapping ofmutantMarker: marker names; N: F2recessive population used in mapping; recombinant: the number of recombinants; B: the structure schematic diagram ofgene. The grey box represents the 5¢un-translated region. The black boxes represent the exons and the horizontal lines represent introns.

(图5)
A: 从左到右分别为突变体、杂交后代中的可育植株、杂交后代中的不育植株、突变体的雄穗, 标尺为10 cm; B: 从左到右分别为突变体、杂交后代中的可育植株、杂交后代中的不育植株、突变体的花药, 标尺为1 mm; C: 从左到右分别为突变体、杂交后代中的可育植株、杂交后代中的不育植株、突变体被I2-KI染色后的花粉粒, 标尺为100 μm。
A: plants from left to right are the tassels of themutant, the fertile hybrid plant, the sterile hybrid plant, and themutant, respectively, bar: 10 cm; B: plants from left to right are the anthers of themutant, the fertile hybrid plant, the sterile hybrid plant, and themutant, respectively, bar: 1 mm; C: plants from left to right are the I2-KI staining of pollen grains of themutant, the fertile hybrid plant, the sterile hybrid plant, and themutant, respectively, bar: 100 μm.
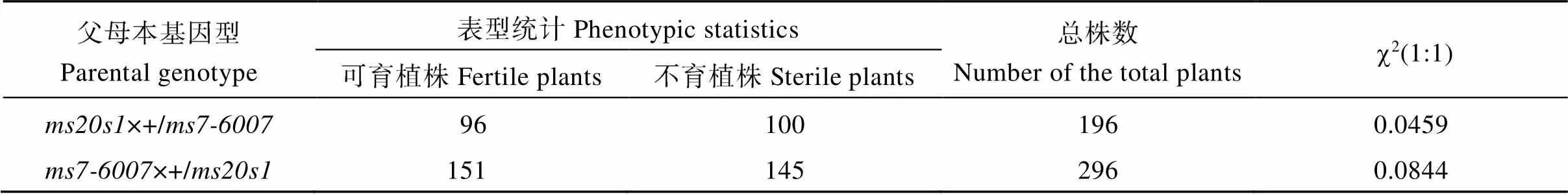
表4 ms20s1与ms7-6007突变体等位测验
c2(0.05) (1)= 3.84.
2.4 ms20s1突变位点的鉴定
为进一步验证确认的突变位点, 以B73中的基因组序列为参考, 共设计3对PCR引物(表1), 分别扩增野生型和突变体中基因序列, 扩增产物覆盖了基因全长。对扩增产物测序发现,突变体中基因在第1个外显子ATG下游22 bp处缺失了3个碱基; 第3个外显子上的第999个碱基由T突变为C, 缬氨酸变为丙氨酸; 第1176个碱基由A突变为G, 组氨酸变为精氨酸; 第1193个碱基由G突变为C, 丙氨酸变为脯氨酸(图4-B)。与已报道的突变体第2外显子有7个碱基插入及突变体第3个外显子有转座子插入的突变方式不同, 以上研究结果充分证实是基因的新等位突变体。
我们根据A到G的SNP变异, 开发了一个CAPS (Cleaved Amplified Polymorphic Sequences)酶切标记。使用引物Ms7-P3 (表1)扩增需要鉴定植株的基因组序列(图6-A),突变体在突变位点附近包含一个B I限制性内切酶的酶切位点, 野生型由于未发生由A到G的变异, 不能被B I酶切(图6-B)。该标记可以作为基因型鉴定的功能性共分离分子标记使用, 为后续不育性状向优异母本种质的回交转育和制种应用奠定基础。
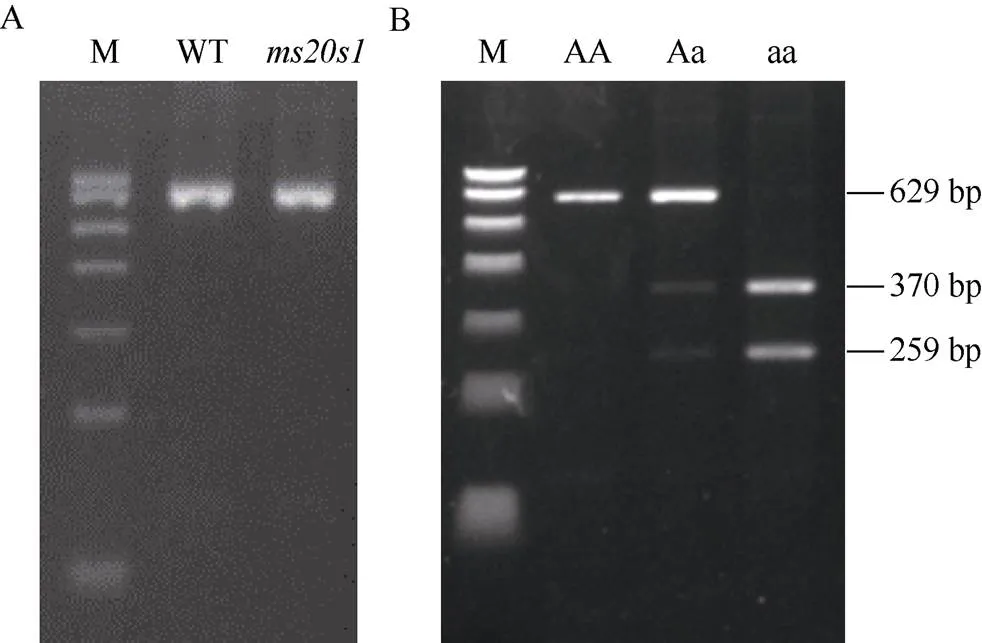
图6 基因型鉴定
A: CAPS标记PCR产物; B:B I酶切产物, AA、Aa、aa分别代表纯合野生型、杂合野生型、纯合突变体。
A: PCR products of the CAPS markers; B:B I digestion product. AA, Aa, and aa represent homozygous wild type, heterozygous wild type, and homozygous mutant, respectively.
2.5 ZmMs7基因的组织特异性分析
突变体主要表现在花药形态及其功能上的改变, 所以我们预测该基因在花药中特异表达。分别提取野生型在苗期, 拔节期和抽雄期不同组织的RNA并反转录, 利用实时荧光定量PCR检测基因的表达量(表1)。结果显示,基因在根、茎、叶等营养器官中的表达量非常微弱, 在花药中特异表达(图7-A)。进一步分析该基因在不同发育阶段花药中的表达水平发现,基因在2.7~3.0 mm长度(S8b~S10)花药中表达, 且在3.0 mm单核小孢子阶段表达量最高(图7-B)[25], 表明基因在四分体期和单核小孢子期的玉米花药发育中具有重要作用[26]。
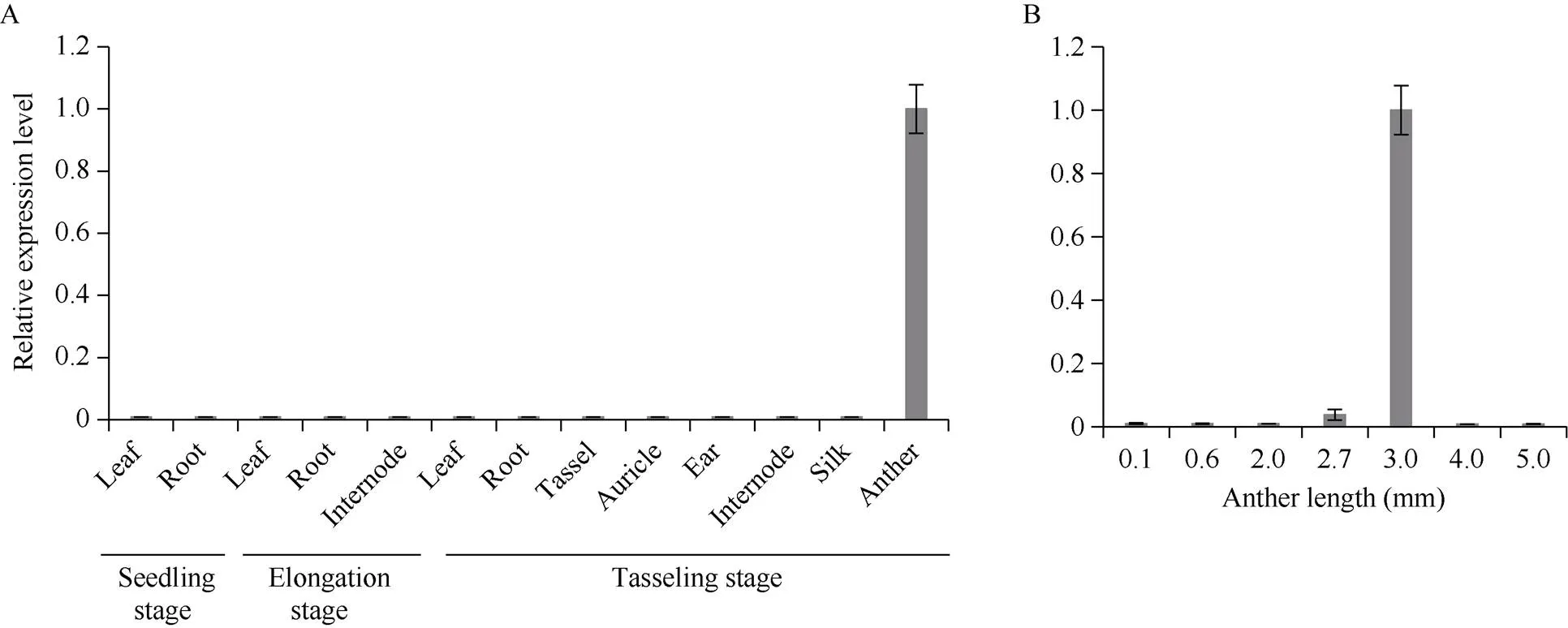
图7 ZmMs7基因在不同组织中的表达量变化
3 讨论
本研究中的属于无花粉型玉米雄性不育突变体。石蜡切片观察分析结果表明,突变体发育异常起始于花药发育的S9阶段, 在S11阶段最为显著, 主要表现为绒毡层细胞降解异常, 小孢子细胞败育, 导致无花粉粒形成。基因定位和等位测验结果表明为已知基因的新等位突变。
玉米基因编码一个PHD-finger的转录因子, 是玉米减数分裂后花药发育的关键调控因子[27]。植物中, PHD是锌指结构域家族的一类转录因子, 参与了胚胎到营养生长, 营养生长到生殖生长中的各项生命过程[28]。在拟南芥、水稻、大麦中已鉴定出该基因的同源基因、/、, 这些基因在雄性育性调控、细胞学表现以及时空表达上也基本相似[29-31]。其中在减数分裂后的绒毡层中特异表达, 参与花粉外壁和花粉胞质组分的形成以及绒毡层的发育,突变体不能进行PCD, 绒毡层细胞异常降解导致线粒体膨胀和大的自噬泡;控制着绒毡层细胞的程序性死亡和功能花粉的发育, 突变体表现为绒毡层细胞增殖失控、细胞膨胀、DNA断裂延迟、乌氏体和花药壁发育异常。这些都表明在植物雄性器官发育过程中PHD-finger蛋白是至关重要的。
已有研究表明,突变体表现出完全隐性雄性不育, 细胞学特征是小孢子壁和芽孔发育不良, 绒毡层细胞发育异常,突变体与其表现相似[27,32]。突变体在减数分裂期小孢子发育正常, 在S11时绒毡层细胞开始肿胀, 小孢子败育, 且在皱缩的花药室中产生很多碎片, 表明绒毡层细胞的程序性死亡降解的异常。在小孢子发育后期,突变体与突变体表现相同。并且突变体的细胞学特征与水稻突变体和拟南芥突变体相似, 表现出绒毡层退化延迟和花粉外壁发育缺陷[33-34]。作为转录激活因子发挥作用, 仅在花药中表达。且在花药发育的四分体期和单核小孢子期特异表达[25]。这与突变体小孢子在四分体期染色体异常浓缩变粗, 绒毡层细胞直至四分体期发育都正常, 但之后绒毡层细胞开始液泡化并迅速降解, 花药小孢子壁和芽孔及绒毡层细胞发育异常这一现象对应[26]。
突变体中基因在第1个外显子上缺失3个碱基, 并没有在重要结构域上。第3个外显子不同位置上有3个单碱基的突变, 引起了3个氨基酸的变化, 且靠近PHD结构域, 可能影响蛋白功能; 而突变体是在第2外显子有7个碱基插入, 导致蛋白翻译发生移码, 造成亮氨酸拉链区域和PHD结构域的缺失; 此外,突变体是在第3外显子上插入了1136个碱基的转座元件, 导致翻译提前终止, 同样造成亮氨酸拉链区和PHD结构域的缺失[27]。由此对比来看,突变体的突变方式与突变体、突变体不同, 证明是基因的一个新等位突变体。我们基于A到G的SNP变异开发了一个CAPS功能标记, 为突变体不育位点向优良杂交种母本回交转育, 以及配套的SPT保持系创制和制种应用奠定了基础。CAPS标记比较直观, 但需要进行酶切和电泳, 检测效率偏低。KASP (Kompetitive Allele Specific PCR)标记检测更为方便, 通量大, 效率高, 后续将在突变位点处开发KASP标记以提高检测效率。
4 结论
本研究发现了一个玉米雄性不育突变体,该突变体表现为绒毡层细胞程序性死亡异常。基因定位结果表明是基因的一个新等位突变体。基因在绒毡层细胞和小孢子的发育过程中特异表达。在突变体中,基因的第3外显子3个碱基的突变造成了编码氨基酸的非同义突变, 可能引起了蛋白功能的改变, 进而造成植株的雄性不育。本研究为隐性核不育系的创制提供了新的突变位点, 同时也为该位点的育种应用提供了有效的功能标记。
[1] Tester M, Langridge P. Breeding technologies to increase crop production in a changing world., 2010, 327: 818–822.
[2] Wan X Y, Wu S W, Li Z W, Dong Z Y, An X L, Ma B, Tian Y H, Li J P. Maize genic male-sterility genes and their applications in hybrid breeding: progress and perspectives., 2019, 12: 321–342.
[3] Chen L, Liu Y G. Male sterility and fertility restoration in crops., 2014, 65: 579–606.
[4] Williams M E. Genetic engineering for pollination control., 1995, 13: 344–349.
[5] Wu Y Z, Fox T W, Trimnell M R, Wang L J, Xu R J, Cigan A M, Huffman G A, Garnaat C W, Hershey H, Albertsen M C. Development of a novel recessive genetic male sterility system for hybrid seed production in maize and other cross-pollinating crops., 2016, 14: 1046–1054.
[6] Zhang D B, Luo X, Zhu L. Cytological analysis and genetic control of rice anther development., 2011, 38: 379–390.
[7] Zhang D B, Wilson Z A. Stamen specification and anther development in rice., 2009, 54: 2342–2353.
[8] Scott R J, Spielman M, Dickinson H G. Stamen structure and function., 2004, 16: S46–S60.
[9] Wang D, Skibbe D S, Walbot V. Maizeexhibits pre-meiotic somatic and post-meiotic microspore and somatic defects but sustains anther growth., 2011, 24: 297–306.
[10] Stieglitz H, Stern H. Regulation of beta-1,3-glucanase activity in developing anthers of Lilium., 1973, 34: 169–173.
[11] Ariizumi T, Toriyama K. Genetic regulation of sporopollenin synthesis and pollen exine development., 2011, 62: 437–460.
[12] Hernandez-Pinzon I, Ross J H E, Barnes K A, Damant A P, Murphy D J. Composition and role of tapetal lipid bodies in the biogenesis of the pollen coat of., 1999, 208: 588–598.
[13] Bih F Y, Wu S S, Ratnayake C, Walling L L, Nothnagel E A, Huang A H C. The predominant protein on the surface of maize pollen is an endoxylanase synthesized by a tapetum mRNA with a long 5' leader., 1999, 274: 22884–22894.
[14] Liu L, Fan X D. Tapetum: regulation and role in sporopollenin biosynthesis in., 2013, 83: 165–175.
[15] Phan H A, Iacuone S, Li S F, Parish R W. The MYB80 Transcription factor is required for pollen development and the regulation of tapetal programmed cell death in., 2011, 23: 2209–2224.
[16] Cui Y, Zhao Q, Xie H T, Wong W S, Wang X F, Gao C J, Ding Y, Tan Y Q, Ueda T, Zhang Y, Jiang L W. MONENSIN SENSITIVITY1 (MON1)/CALCIUM CAFFEINE ZINC SENSITIVITY1 (CCZ1)-mediated rab7 activation regulates tapetal programmed cell death and pollen development., 2017, 173: 206–218.
[17] Aya K, Ueguchi-Tanaka M, Kondo M, Hamada K, Yano K, Nishimura M, Matsuoka M. Gibberellin modulates anther development in ricethe transcriptional regulation of GAMYB., 2009, 21: 1453–1472.
[18] Skibbe D S, Wang X J, Borsuk L A, Ashlock D A, Nettleton D, Schnable P S.Floret-specific differences in gene expression and support for the hypothesis that tapetal degeneration ofLoccursprogrammed cell death., 2008, 35: 603–616.
[19] Zhao D Z, Wang G F, Speal B, Ma H. Thegene encodes a putative leucine-rich repeat receptor protein kinase that controls somatic and reproductive cell fates in theanther., 2002, 16: 2021–2031.
[20] Fu Z Z, Yu J, Cheng X W, Zong X, Xu J, Chen M J, Li Z Y, Zhang D B, Liang W Q. The rice basic helix-loop-helix transcription factor TDR INTERACTING PROTEIN2 is a central switch in early anther development., 2014, 26: 1512–1524.
[21] Moon J, Skibbe D, Timofejeva L, Wang C J R, Kelliher T, Kremling K, Walbot V, Cande W Z. Regulation of cell divisions and differentiation by MALE STERILITY32 is required for anther development in maize., 2013, 76: 592–602.
[22] Guo Z F, Wang H W, Tao J J, Ren Y H, Xu C, Wu K S, Zou C, Zhang J N, Xu Y B. Development of multiple SNP marker panels affordable to breeders through genotyping by target sequencing (GBTS) in maize., 2019, 39: 37.
[23] Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R. Genome sequencing reveals agronomically important loci in rice using MutMap., 2012, 30: 174–178.
[24] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) method., 2001, 25: 402–408.
[25] Han Y J, Hu M J, Ma X X, Yan G, Wang C Y, Jiang S Q, Lai J S, Zhang M. Exploring key developmental phases and phase- specific genes across the entirety of anther development in maize., 2022, 64: 1394–1410.
[26] An X L, Ba B, Duan M J, Dong Z Y, Liu R G, Yuan D Y, Hou Q C, Wu S W, Zhang D F, Liu D C, Yu D, Zhang Y W, Xie K, Zhu T T, Li Z W, Zhang S M, Tian Y H, Liu C, Li J P, Yuan L P, Wan X Y. Molecular regulation ofrequired for maize male fertility and development of a dominant male-sterility system in multiple species., 2020, 117: 23499–23509.
[27] Zhang D F, Wu S W, An X L, Xie K, Dong Z Y, Zhou Y, Xu L W, Fang W, Liu S S, Liu S S, Zhu T T, Li J P, Rao L Q, Zhao J R, Wan X Y. Construction of a multicontrol sterility system for a maize male-sterile line and hybrid seed production based on thegene encoding a PHD-finger transcription factor., 2018, 16: 459–471.
[28] Halbach T, Scheer N, Werr W.Transcriptional activation by the PHD finger is inhibited through an adjacent leucine zipper that binds 14-3-3 proteins., 2000, 28: 3542–3550.
[29] Yang C, Vizcay-Barrena G, Conner K, Wilson Z A. MALE STERILITY1 is required for tapetal development and pollen wall biosynthesis., 2007, 19: 3530–3548.
[30] Ito T, Shinozaki K. Thegene of, encoding a nuclear protein with a PHD-finger motif, is expressed in tapetal cells and is required for pollen maturation., 2002, 43: 1285–1292.
[31] Gomez J F, Wilson Z A. A barley PHD finger transcription factor that confers male sterility by affecting tapetal development., 2014, 12: 765–777.
[32] Morton C M, Lawson D L, Bedinger P. Morphological study of the maize male sterile mutant., 1989, 34: 239–245.
[33] Li H, Yuan Z, Vizcay-Barrena G, Yang C Y, Liang W Q, Zong J, Wilson Z A, Zhang D B.encodes a PHD-finger protein that is required for tapetal cell death and pollen development in rice., 2011, 156: 615–630.
[34] Yan X J, Ma L, Pang H Y, Wang P, Liu L, Cheng Y X, Cheng J K, Guo Y, Li Q Z. METHIONINE SYNTHASE1 is involved in chromatin silencing by maintaining DNA and histone methylation., 2019, 181: 249–261.
Genetic analysis and molecular identification of a multiple allele mutant ofgene in maize
CAO Xiao-Xiong1,2, LIU Yi-Fan1,2, ZHOU Yu-Qiang2, WANG Jing2, WU Yu-Jin2, WANG Hong-Wu2, LI Kun2, LIU Xiao-Gang2, HUANG Chang-Ling2, LIU Zhi-Fang2, GUO Jin-Jie1,*, and HU Xiao-Jiao2,*
1College of Agronomy, Hebei Sub-center of National Maize Improvement Center of China / State Key Laboratory of North China Crop Improvement and Regulation / Hebei Agricultural University, Baoding 071001, Hebei, China;2Institute of Crop Sciences, Chinese Academy of Agricultural Sciences / National Engineering Research Center of Crop Molecular Breeding, Beijing 100081, China
We identified a maize male sterile mutant in the natural population, designated as. The mutant had complete male sterility and lacked pollen grains in the withered anthers. Cytological analysis showed that, compared with the wild type, themutant exhibited shrinkage locule, swollen tapetum cells, and aborted microspore at S11 stage, indicating that themutant had abnormal tapetal programmed cell death and complete pollen abortion. Genetic analysis revealed that the male sterility trait was controlled by a single recessive nuclear gene. To clone the target gene, we constructed the F2populations by crossingwith different inbred lines and analyzed the population genotype using genotyping by target sequencing (GBTS) technology. The gene was initially mapped to the 124.95–128.47 Mb region on chromosome 7, and the interval was narrowed down to 0.68 Mb after fine mapping. Bioinformatics analysis indicated that there was one known genein this region.Thegene encoded a PHD-finger transcription factor that played an important role in tapetum development and pollen wall formation. Allelism test demonstrated thatwas an allelic mutant ofgene. Gene sequencing results showed that themutant had multiple sequence variants in the exon region, which were different from the reported mutantsand, confirmingwas a new allelic mutant of. The discovery and identification of themutant provide a new material for exploring the molecular mechanism and breeding application of maize genic male sterility.
maize;;; gene mapping; allelic mutant
10.3724/SP.J.1006.2023.33002
本研究由国家重点研发计划项目(2022YFD1200802),中国农业科学院科技创新工程(CAAS-ZDRW202004),作物分子育种国家工程研究中心和河北省玉米现代种业科技创新团队项目(21326319D)资助。
This study was supported by the National Key Research and Development Program of China (2022YFD1200802), the Agricultural Science and Technology Innovation Program (CAAS-ZDRW202004), the National Engineering Research Center of Crop Molecular Breeding, and the Science and Technology Innovation Team of Maize Modern Seed Industry in Hebei (21326319D).
胡小娇, E-mail: huxiaojiao@caas.cn; 郭晋杰, E-mail: guojinjie512@163.com
E-mail: caoxx13@163.com
2023-01-05;
2023-04-17;
2023-05-05.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20230504.1553.002.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
作物学报(2022年6期)2022-04-08 01:26:24
湖北农业科学(2021年19期)2021-11-15 07:01:38
国际医学放射学杂志(2021年5期)2021-10-22 07:26:20
云南农业大学学报(自然科学)(2021年3期)2021-06-11 04:19:38
麦类作物学报(2018年4期)2018-05-11 09:34:08
第一财经(2017年36期)2017-09-25 06:17:11
广西植物(2016年10期)2016-11-11 06:51:39
西南农业学报(2016年4期)2016-05-17 05:41:45
中国瓜菜(2014年3期)2014-04-29 00:44:03
中国蔬菜(2013年8期)2013-01-28 04:52:48
