
菊粉改性含氢硅油表面活性剂的合成及性能研究
2023-09-13 06:19张万强陈洪龄
精细石油化工进展 2023年4期
张万强,陈洪龄
南京工业大学化工学院,江苏 南京 211816
菊粉(IN)又称菊糖,是一种天然无污染、储藏丰富、可再生的多聚果糖,主要来源于芦荟、菊苣、菊芋等植物。菊粉是由D-呋喃果糖分子经β(1-2)糖苷键相连而成的直链果糖,分子链末端以连接一个葡萄糖残基[1]而结束。由于菊粉具有优异的增稠性、溶解性、吸湿性、稳定性及药理活性等,使其在食品、医药和日用化工等领域得到广泛应用[2-4]。菊粉果糖单元含有丰富的羟基,使菊粉具有相当高的化学活性,能够进行酯化、醚化及羧甲基化等化学反应。
由于石油等化石资源具有不可再生性且对环境污染严重,因此菊粉凭借其可生物降解、对环境无污染、储量大及可再生等优异性能完全符合可持续发展理念,已经出现逐步取代依赖传统石化来源的表面活性剂的趋势。Jordi 等[5]使用KOH作为催化剂,对菊粉进行疏水改性,制备了菊粉羟烷基醚,并研究了疏水基团长度、果糖单元接枝基团数、反应温度、溶剂异丙醇用量、不同碱性催化剂及用量对反应效率的影响。韩乐等[6]利用Ce(NH4)2(NO3)6作为引发剂,采用水热合成法成功制备了丙烯酰胺型菊粉,并考察了引发剂浓度、反应时间、反应温度、丙烯酰胺浓度对反应转化率和接枝率的影响,研究表明产物具备良好的絮凝效果。除此之外,由于菊粉中含有大量的亲水性羟基,可利用其具有强疏水性、良好的稳定性、优异的界面性等特性的聚合物含氢硅油,选择性地对菊粉进行疏水改性,通过调节菊粉中反应羟基数量和含氢硅油的含氢量,来控制产物的亲水亲油平衡值(HLB),获得具有良好乳化性能的表面活性剂[7]。
本研究利用烯丙基缩水甘油醚(AGE)与含氢硅油进行硅氢加成反应,使长硅链引入环氧基,然后再用菊粉中的羟基与环氧基反应,对含氢硅油进行亲水改性,制备了菊粉改性含氢硅油表面活性剂。通过调节菊粉与含氢硅油的摩尔比制备不同HLB 的产物,通过傅里叶变换红外光谱仪对产物进行结构表征,并测试产物的表面张力、接触角、泡沫性能和乳化性能,以期为菊粉的应用提供数据。
1 材料与方法
1.1 试剂和仪器
菊粉(质量分数>98%),山东鲁森生物科技有限公司;烯丙基缩水甘油醚(AGE),分析纯,上海阿拉丁生化科技股份有限公司;0.5%含氢硅油(0.5%PHMS,其中H 质量分数为0.5%)、烷基糖苷(APG 0814,工业级),南京盛泰克新材料有限公司;氯铂酸、异丙醇、钠型732 阳离子交换树脂,分析纯,国药集团化学试剂有限公司;NaOH,分析纯,西陇化工股份有限公司;N,N-二甲基甲酰胺,分析纯,上海凌峰化学试剂有限公司;实验室用水均为去离子水,自制。
SL200B 型静态接触角检测仪,上海梭伦信息科技有限公司;WQF-510A 型傅里叶变换红外光谱仪,北京北分瑞利分析仪器有限公司;BZY系列自动表面张力仪,上海方瑞仪器有限公司。
1.2 实验方法
准确称量含0.025 mol Si—H 的0.5% PHMS、0.03 mol AGE 和质量分数为30%的异丙醇溶液,加入配有搅拌和加热的三口烧瓶,超声混合均匀,开启冷凝水,通入N2,温度升至80 ℃,向三口烧瓶中加入质量分数为0.003%的氯铂酸催化剂,维持80 ℃反应4 h,然后100 ℃减压旋转蒸馏除去异丙醇溶剂和未反应的AGE,得到中间体AGE-0.5%PHMS。
将AGE-0.5%PHMS 与5 gN,N-二甲基甲酰胺、10 g 异丙醇超声混合,加入恒压漏斗中。向装有冷凝管、恒压漏斗的三口烧瓶中加入4.05 g(0.025 mol)菊粉(162 g/mol)、20 g 去离子水和0.15 g NaOH,常温搅拌30 min 后升温至90 ℃,开启恒压漏斗缓慢滴加,恒温反应4 h。反应结束后加入阳离子树脂调节pH 为中性,抽滤除去溶液中的树脂,然后将滤液100 ℃减压旋转蒸馏除去溶剂,得到菊粉改性含氢硅油表面活性剂(IN-0.5% PHMS)。通过调节原料的摩尔比(菊粉与Si—H 的摩尔比分别为1∶1、2∶1、3∶1),制备了3种菊粉改性含氢硅油表面活性剂,反应式见式(1)和(2)。
1.3 表征与测试
1.3.1 红外光谱分析
将适量的样品和KBr粉末用压片机加压制备成薄片,使用傅里叶变换红外光谱仪(FT-IR)对样品进行表征。
1.3.2 表面张力的测定
参照文献[8-9]相关方法测试产物的表面张力。取0.5 g 样品于适量去离子水中溶解,将溶液转移到250 mL 容量瓶中定容,配制成2 g/L 的标准溶液。取适量标准溶液,加去离子水稀释成不同浓度的溶液,再将溶液在25 ℃恒温水浴锅中恒温2 h,随后用表面张力仪测试不同浓度溶液的表面张力。
1.3.3 接触角测定
配制1 g/L 的表面活性剂溶液并放置于25 ℃恒温水浴锅中,恒温2 h。用取样针取2.5 mL 溶液,滴一滴在光滑的固体石蜡片上,测量2 min 内液滴在固体石蜡片上的接触角。
1.3.4 泡沫性能测定
参照文献[10]相关方法测试产物的泡沫性能。配制2 g/L 的表面活性剂溶液并在25 ℃恒温水浴锅中恒温静置2 h。取20 mL 待测表面活性剂溶液置于100 mL 具塞量筒中,上下振动40 次,静置后观察泡沫体积变化,记录初始泡沫体积(V0)、1 min时的泡沫体积(V1)和5 min时的泡沫体积(V2),并计算5 min 时的消泡率(η),具体计算见式(3)。
1.3.5 乳化性能测定
参照文献[11-13]相关方法测试表面活性剂的乳化性能。向50 mL离心管中加入0.1 g菊粉改性含氢硅油表面活性剂、7 g去离子水和3 g油相,通过超声波细胞粉碎机乳化3 个周期(每个周期工作25 s、间隔25 s)制备乳液,然后用手电筒从乳液背部照射观察乳液状态并记录乳液开始分层的时间。乳液未分层时间越长,稳定性越好。
乳液静置稳定性:将乳液放置在25 ℃室温环境下,观察乳液状态。
乳液耐热稳定性:将乳液放置在恒温恒湿箱中,调至50 ℃,8:30 开烘箱,22:30 关烘箱,观察乳液状态。
乳液耐冷稳定性:将乳液放置在5 ℃冰箱中,观察乳液状态。
乳液离心稳定性:将乳液放置于台式高速离心机中,调节转数为3 000 r/min、离心10 min,观察乳液状态。
为了研究菊粉改性含氢硅油与其他绿色表面活性剂的复配乳化性能,选择烷基糖苷(APG 0814)和菊粉改性含氢硅油复配作为乳化剂。
复配乳液制备:离心管中加入0.1 g APG 0814、0.1 g 菊粉改性含氢硅油、3 g 二甲基硅油(350 Pa·s)、7 g 水,超声波细胞粉碎机乳化3 个周期制备乳液。
2 结果与讨论
2.1 FT-IR测试结果
图1 为0.5%PHMS、IN、AGE、中间体AGE-0.5%PHMS 和IN-0.5%PHMS 的红外光谱。由图1可知:0.5% PHMS 中2 165 cm-1处为Si—H 的伸缩振动峰,1 260 cm-1处为Si—CH3的对称变形振动特征峰,800 cm-1处为Si—C 的不对称伸缩振动峰,1 080 cm-1处为Si—O—Si 键的反对称伸缩振动峰。AGE中1 650 cm-1处为C=C键的伸缩振动峰,3 055 cm-1处为环氧基中—CH 的伸缩振动峰,1 255 cm-1处为环氧基的对称伸缩振动峰,910 cm-1处为环氧基的反对称伸缩振动吸收峰[14]。AGE-0.5% PHMS 中2 970 cm-1处为—CH3的反对称伸缩振动峰,2 910 cm-1处为—CH2的反对称伸缩振动峰,1 650 和2 165 cm-1处C=C 键和Si—H键的特征峰消失且环氧基的特征峰存在,表明中间体制备成功。IN-0.5% PHMS中3 300 cm-1处存在—OH 的红外特征吸收峰,1 260、800、1 080、2 970、2 910 cm-1处的红外特征峰证明产物中存在长硅链,同时3 055、910 cm-1处环氧基的红外特征峰消失且1 260 cm-1处环氧基的对称伸缩振动峰明显降低。综上可知,最终制备的产物为菊粉改性含氢硅油。
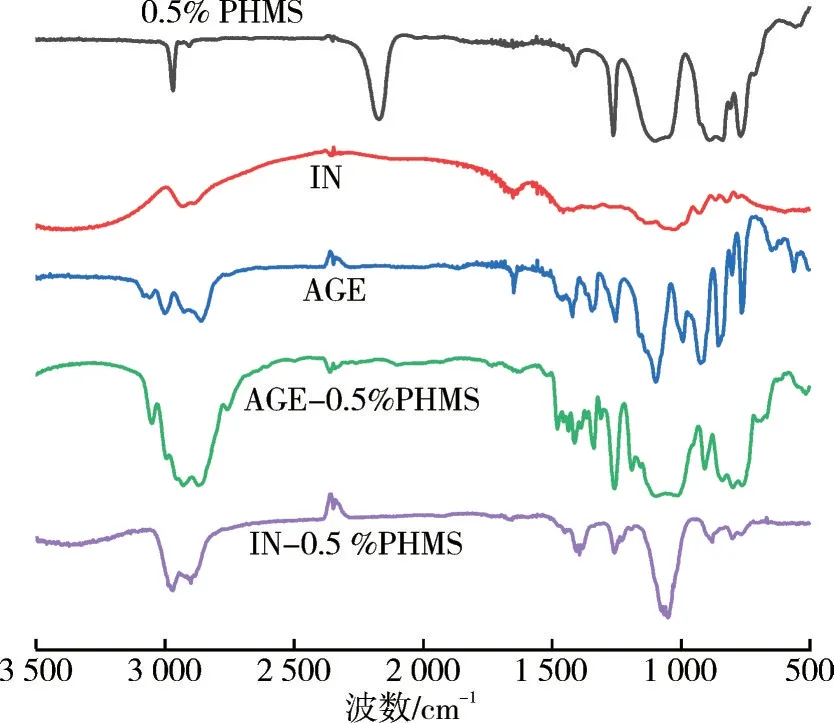
图1 样品的红外光谱
2.2 表面张力性能分析
图2 为IN-0.5%PHMS 表面活性剂溶液的表面张力(γ)随溶液浓度变化曲线。由图2可知:随着IN-0.5%PHMS水溶液质量浓度的增加,溶液的表面张力呈先迅速下降、然后缓慢降低、最终趋于平衡的趋势;随着菊粉原料比的增加,产物临界胶束浓度增加,最低表面张力降低,其中原料比为3∶1 时,产物降低表面张力能力最佳,产物水溶液最低表面张力为26.17 mN/m,临界胶束浓度为0.15 g/L,这是因为原料中菊粉含量增加,产物亲水端支链化程度增加,产物形成胶束难度增加,产物聚集溶液表面趋势增加,临界胶束浓度增加,溶液最低表面张力降低。可见,IN-0.5%PHMS 表面活性剂保留了0.5%PHMS 良好表面活性的优点,具备较低的表面张力。

图2 IN-0.5%PHMS表面活性剂溶液表面张力随质量浓度的变化曲线
2.3 接触角性能测试
将IN-0.5%PHMS 表面活性剂配制成1 g/L 的表面活性剂水溶液,测量其在光滑固体石蜡片上接触角随时间的变化。
图3 为不同原料比IN-0.5%PHMS 表面活性剂在固体石蜡表面的接触角随时间变化的曲线。由图3 可知:IN-0.5%PHMS 表面活性剂在石蜡片上的接触角随着时间延长而逐渐降低,最后到达一个稳定值;随着原料中菊粉比例增加,表面活性剂的亲水性增强,在石蜡片表面的初始接触角和平衡接触角变大,其中初始接触角由83.55°增到94.90°,平衡接触角由71.38°增到78.33°。

图3 IN-0.5%PHMS表面活性剂在固体石蜡片表面接触角随时间变化曲线
2.4 泡沫性能测试
有机硅表面活性剂溶液具有较低表面张力,不溶性气体借助外力作用进入液体中形成具有一层薄液膜包裹的气泡。如果液体为纯水,气泡到达液体表面,表面张力较大,气泡容易破裂;而表面活性剂一方面能降低水的表面张力,另一方面又能在气液界面进行吸附,增加气泡的液膜强度,从而容易在液体表面形成泡沫。所以,泡沫性能也是有机硅表面活性剂重要性能之一。
表1 为各表面活性剂溶液的初始(V0)、1 min(V1)、5 min(V2)的泡沫体积和5 min时自身消泡率(η)。由表1可知:各产物水溶液的初始气泡高度几乎相同,随着菊粉原料比的增加,产物亲水端支链化程度增加,表面活性剂分子在气液界面吸附不够紧密,吸附膜强度降低,泡沫稳定性降低,自身消泡率增加。其中原料比为 2∶1时产物起泡性最好、消泡率最高,初始泡沫体积为20 mL,5 min自身消泡率为70.0%。
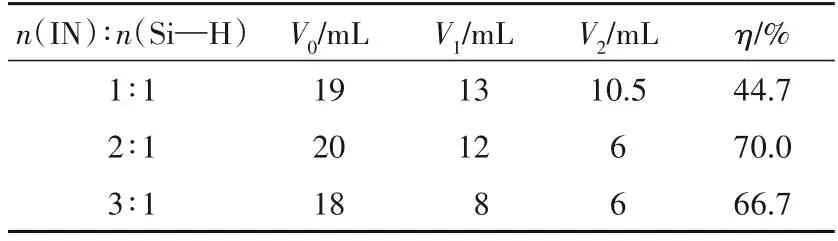
表1 泡沫性能测试
2.5 乳化性能测试
由于IN-0.5%PHMS 表面活性剂中同时含有亲水性的—OH 和亲油性的Si—O—Si 链,因此具备双亲性结构,能够在原本互不相溶的油水两相界面处进行吸附,使其中一相均匀地分散在另一相中,得到均相的乳液。
表2为IN-0.5%PHMS表面活性剂乳化4种油相的分层时间。图4 为不同原料比IN-0.5%PHMS 表面活性剂乳化4 种油相制备乳液的状态。图5为原料比1∶1制备的IN-0.5%PHMS乳化4 种油相制得乳液的微观形貌。由表2 和图4~5 可知:IN-0.5%PHMS 表面活性剂均能够对4 种油相乳化形成乳液。其中,原料比为1∶1 时乳化效果最好,乳化液体石蜡乳液在室温下放置20 d未分层,对二甲基硅油、环己烷、正辛烷乳化所形成的乳液静置2 d均出现分层,这是因为二甲基硅油(350 Pa·s)黏度大,乳化时油相难以形成尺寸较小且均匀的乳液,而环己烷和正辛烷属于烷烃,密度与水差异大,乳化后形成液滴较小,布朗运动影响明显,液滴之间容易相互碰撞并发生聚合现象,所以稳定性相对较差。

表2 IN-0.5%PHMS乳化不同油相分层情况

图4 IN-0.5% PHMS乳化不同油相的状态

图5 IN-0.5% PHMS 1∶1乳化不同油相所得乳液微观形貌
由于原料比为1∶1 制备的IN-0.5%PHMS 乳化液体石蜡所制备的乳液稳定性较好,对该乳液的乳液稳定性进一步测试,结果如表3 所示。由表3 可知:该乳液室温下20 d 未分层,耐热稳定性测试4 d后开始分层,耐冷稳定性测试14 d后乳液底部开始分水分层,3 000 r/min 离心10 min 后,乳液发生黏壁分层现象。

表3 乳液稳定性测试
将原料比为1∶1 产物与烷基糖苷APG 0814进行复配,乳化二甲基硅油(350 Pa·s),考察乳化剂的复配乳化性能,结果如表4 所示。由表4 可知:复配后乳液在室温下放置40 h 后乳液才开始出现分层,而单一IN-0.5%PHMS乳化剂制备乳液时室温静置24 h后开始分层,单一APG 0814乳化剂制备乳液时室温静置35 h 后开始分层,对比可知,复配后乳液稳定性明显增加。此外,复配后乳液的耐热稳定性和耐冷稳定性均比单一APG 乳化剂制备乳液的稳定性有明显提升。复配后乳液的离心稳定性提升不大,均发生分层现象。

表4 复配乳液稳定性
图6 为原料比1∶1 的菊粉改性含氢硅油与APG 0814 复配乳化剂和单一APG 0814 乳化剂乳化二甲基硅油(350 Pa·s)乳液微观形貌。由图6可知:复配后乳液液滴尺寸相对较小且分布均匀,有利于乳液的稳定。

图6 复配乳化剂和APG 0814乳化二甲基硅油(350 Pa·s)乳液微观形貌
3 结论
1)以菊粉和含氢硅油为原料,制备了3 种不同原料比的表面活性剂。通过傅里叶变换红外光谱仪对产物进行结构表征,表明合成产物即为目标产物菊粉改性含氢硅油表面活性剂。
2)菊粉改性含氢硅油表面活性剂具备良好的界面性能,能够明显降低水的表面张力,其中n(IN)∶n(Si—H)为 3∶1 时产物降低表面张力效果最好,其临界胶束浓度为0.15 g/L,最低表面张力为26.17 mN/m。
3)菊粉改性含氢硅油表面活性剂溶液在固体石蜡片上具有良好的铺展能力,其中n(IN)∶n(Si—H)为1∶1 时铺展性能最好,其初始接触角为83.55 °,平衡接触角为71.38 °。
4)菊粉改性含氢硅油表面活性剂具有良好的自身泡沫消泡率,当n(IN)∶n(Si—H)为2∶1时,产物5 min时自身消泡率可达70.0%。
5)菊粉改性含氢硅油表面活性剂对二甲基硅油(350 Pa·s)、液体石蜡、环己烷、正辛烷4 种油相均能乳化,当n(IN)∶n(Si—H)为1∶1 时产物乳化效果最好,其乳化液体石蜡所得乳液室温下静置20 d 未分层,且耐热、耐冷、离心稳定性良好。将IN-0.5% PHMS (原料比为1∶1)与APG 0814 乳化剂复配后乳化二甲基硅油(350 Pa·s),乳液液滴尺寸明显降低,乳液稳定性有明显提升。
猜你喜欢
上海计量测试(2020年1期)2020-03-18
陶瓷学报(2019年5期)2019-01-12
百科知识(2018年22期)2018-11-27
饮食科学(2016年9期)2016-11-18
重庆工商大学学报(自然科学版)(2015年10期)2015-12-28
中国塑料(2015年1期)2015-10-14
当代化工研究(2015年1期)2015-02-20
汽车零部件(2014年5期)2014-11-11
自动化博览(2014年9期)2014-02-28
丝绸(2014年12期)2014-02-28
