
联合策略优化葡萄糖氧化酶的酵母表达
2023-08-30 01:57:12罗同阳王庆庆高庆华王云鹏唐兆宏耿革霞
河北省科学院学报 2023年4期
王 玥,董 聪,罗同阳,王庆庆,高庆华,王云鹏,唐兆宏,2,耿革霞
(1.河北省微生物研究所有限公司,河北 保定 071051;2.保定鲜尔康生物工程有限责任公司,河北 保定 071051;3.河北省药品职业化检查员总队,河北 石家庄 050024)
葡萄糖氧化酶(Glucose oxidase,EC1.1.3.4,简称GOD)是一种氧化还原酶,被广泛应用于食品[1,2]、生物医疗[3]、饲料[4,5]等领域,是生物行业中重要工具用酶之一。目前面对市场的大量需求,寻找高酶活且稳定性好的葡萄糖氧化酶迫在眉睫。
基因工程技术的发展既能实现其高效表达,也能改良其稳定性。GOD 已被应用于多种表达系统中表达。刘春莹等人[6]从海洋细菌中克隆葡萄糖氧化酶基因连接到表达载体 pET28a(+) 中,该重组载体在大肠杆菌 E.coli BL21(DE3) 中被成功表达,测定酶活为 2.04 U/mL。然而,通过原核表达系统表达的外源蛋白,常以二聚体的形式存在,一般没有生物活性且酶活低,因此,目前 GOD 的异源表达多采用真核表达系统,特别是巴斯德毕赤酵母(Pichiapastoris)表达系统。毕赤酵母表达系统实现外源蛋白高通量表达的研究一直受到大量关注[7,8],GOD 在毕赤酵母表达体系中高效表达的策略有很多,如表达元件优化,密码子优化,改造蛋白的分泌途径,发酵条件优化等[9],近年来共表达分子伴侣[10]和增加基因拷贝数[11]是实现GOD高表达的有效手段。而单因素优化后蛋白分泌表达水平的提升不太明显,利用组合优化策略更能促进蛋白修饰,提高分泌水平,获得高酶活的重组菌株[10,12,13]。Yu 等人[12]通过多拷贝筛选、共表达UPR激活因子(Hac1p)构建出 GOD 高产菌株,并在1 L发酵罐(BMGY培养基)中优化GOD 酶活高达2 125.3 U/mL,是目前文献报道中的最高水平。
我们在前期工作中,构建了共表达分子伴侣PDI-Ero1的工程菌株X33/pMD-GOD/pPICZ-PDI-Ero1,在10 L发酵罐中优化诱导策略GOD 酶活为 736 U/mL[14],同时还构建了表达载体pMD-AOX(含AOX 强启动子和终止子,G418 抗性基因)、分子伴侣重组质粒( pPICZB-HAC1n(n=1,2,3或4),pPICZB-PDIn(n=1,2,3或4))。在此基础上,本文采用组合优化的策略提高GOD在毕赤酵母中的表达,首先分别构建了重组表达载体 pMD-AOX-GOD和多拷贝表达载体 pPICZαA-GODn(n=1-5),通过高浓度G418、博来霉素抗性筛选平板,得到 1株高表达GOD的重组菌。在此基础上,分别构建了HAC1、PDI 两种分子伴侣1-4拷贝共表达重组菌株,进行高密度发酵,提高了GOD的高效表达和积累,对促进其工业化大规模生产具有重要意义。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
黑曲霉(Aspergillusniger)菌株、毕赤酵母(Pichiapastoris)X33均由河北省微生物研究所有限公司保存;大肠杆菌EscherichiacoliTOP10 菌株购自天根生化科技有限公司;表达载体pMD-AOX(含AOX 强启动子和终止子,G418 抗性基因)、分子伴侣重组质粒pPICZB-HAC1n(n=1,2,3或4),pPICZB-PDIn(n=1,2,3或4)均由本实验室构建;表达载体pPICZαA购自优宝生物公司。
1.1.2 试剂与培养基
DNA聚合酶、连接酶、限制性内切酶、虾碱性磷酸酶rSAP等工具酶均购自NEB公司;其他试剂均为国产或进口分析纯;PCR产物纯化试剂盒、质粒提取试剂盒均购自全式金生物科技公司;遗传霉素(G418)、博来霉素(Zeocin) 均购自索莱宝生物科技有限公司;酵母基因组DNA提取试剂盒、DNA凝胶回收试剂盒均购自北京天根生物技术有限公司;引物合成和测序工作由北京中科希林生物科技有限公司完成(表1)。LB、MD、YPD、BMGY、YPCS、YPDS等培养基参照Invitrogen公司的毕赤酵母表达操作手册配制。

表1 本文中所用的引物
1.2 实验方法
1.2.1 表达载体pMD-AOX-GOD的构建及毕赤酵母转化
采用酵母基因组DNA提取试剂盒方法提取黑曲霉的基因组DNA。根据表1引物进行葡萄糖氧化酶基因的克隆,PCR反应条件:98 ℃ 5 min;98 ℃ 30 s,65 ℃ 60 s,72 ℃ 10 min,共30个循环;72 ℃ 10 min;16 ℃ 30 min。PCR 产物经纯化回收后采用 GIBSON 方法连接到表达载体pMD-AOX中XhoⅠ、NotⅠ酶切位点上,并转化至大肠杆菌E.coliTOP10。酶切验证筛选出阳性克隆送测序,获得质粒pMD-AOX-GOD。
将表达载体 pMD-AOX-GOD经PmeⅠ酶切线性化并回收后,电击转化至毕赤酵母感受态 X33 中,涂板于 MD 平板上,30 ℃培养2-3 d。用牙签将MD平板生长的单菌落,分别点种至含 G418 梯度( 0.10、0.20、0.50、1.00、2.00 mg/mL) 的YPDS 平板上,30 ℃培养 3-5 d 后,观察菌落生长情况,筛选重组菌株。
1.2.2 多拷贝表达载体的构建及毕赤酵母转化
根据表1引物进行葡萄糖氧化酶基因的克隆,PCR反应条件:98 ℃ 5 min;98 ℃ 30 s,62 ℃ 60 s,72 ℃ 10 min,共20个循环;72 ℃ 10 min;16 ℃ 30 min。PCR 产物经纯化回收后采用 GIBSON 方法连接到表达载体pPICZαA中KpnⅠ、NotⅠ位点上,并转化到E.coliTOP10。酶切验证筛选出阳性克隆送去测序,获得质粒pPICZαA-GOD。
表达载体pPICZαA 的上游有BglⅡ 酶切位点,下游有BamHⅠ酶切位点,该载体经BglⅡ/BamHⅠ双酶切后会产生互补的粘性末端。利用BglⅡ、BamHⅠ 消化阳性载体,两个表达框正向连接从而无法被切开,多拷贝表达载体即可再插入到BamHⅠ 酶切位点,重复插入含有5′ AOX1、目的片段和AOX1 TT 的表达框,由此来构建多拷贝表达载体,如图1所示。

图1 多拷贝表达盒载体的构建流程图
将多拷贝表达载体利用BamHⅠ 酶切线性化,经回收后分别电转化至毕赤酵母X33感受态中,涂板于含有 100 μg/mL Zeocin 抗性的YPDS平板上,30 ℃ 培养 2-3 d 筛选含多拷贝基因的重组菌株。
1.2.3 重组菌株的筛选
利用1.2.1和1.2.2方法获得的转化子点种至显色筛选MM平板(Tryptone 20.0 g/L,Yeast extract 10.0 g/L,1%甲醇,2%琼脂)上,30 ℃培养1 d,取出平板倒入显色反应液(1% 邻联茴香胺溶液、3% 的90 U/mL辣根过氧化物酶、10%的18%葡萄糖溶液,1%琼脂),30 ℃ 放置2 h后观察显色圈,筛选出棕色圈颜色深、面积大的菌株用于进一步试验。将上述菌株接种于5 mL YPCS液体培养基中,30℃摇床培养20~24 h后OD600为10 左右,加入1% 甲醇,此后每隔24 h加入1% 甲醇诱导,诱导72 h后收菌,离心收集上清液,测定酶活。比较两种方法,选择酶活相对高且稳定性较好的菌株作为第一代高产菌株(X33/AnGOD),并制备成感受态备用。
将分子伴侣HAC1,PDI多拷贝表达载体分别用BamHⅠ酶切线性化,电转化至出发菌X33/AnGOD中,涂布于YPDS抗性筛选平板上,获得阳性转化子,分别命名为X33/AnGOD-HAC1n(n=1,2,3或4),X33/AnGOD-PDIn(n=1,2,3或4),试管水平测定GOD酶活。
1.2.4 重组表达菌株中基因拷贝数的检测
荧光定量 PCR (RT-qPCR)方法参照 Takara 公司试剂盒(SYBR®Premix ExTaqTMkit)。仪器使用实时荧光定量PCR仪(Thermofisher QuantStutio 1),PCR 反应条件为: 50 ℃ 2 min,95 ℃ 10 min 后开始循环; 95 ℃ 15 s,60 ℃ 20 s,72 ℃ 40 s,共40个循环。以GAPDH为内参基因,采用比较Ct值法(2-(ΔΔCt)法)分析确定重组菌株所含的基因拷贝数。使用的 RT-qPCR 引物见表1。
1.2.5 10 L 发酵罐高密度发酵
将采用组合优化策略筛选出的重组菌株X33/AnGOD-PDI划线培养。挑取单菌落接种于5 mL YPD 液体培养基中,30 ℃培养12 h后按3%的接种量转接至150 mL BMGY 培养基中,30 ℃培养至菌液OD600≈8~10接种于发酵罐中。10 L发酵罐中含有4.5 L BSM 培养基,发酵罐灭菌后以10% 发酵体积(5 L)进行接种,采用浓氨水自动流加以控制发酵期间 pH 值,发酵温度设置为30 ℃。具体发酵过程分为菌体培养阶段、碳源饲喂阶段、饥饿培养阶段和诱导表达阶段等4个阶段,具体方法参见Invitrogen手册。每隔12 h 检测OD600、GOD活性,同时采用SDS-PAGE 检测表达量的累积。
1.2.6 葡萄糖氧化酶的酶活测定
GOD活性测定方法参照文献[14],采用邻-联(二)茴香胺分光光度法。
2 结果与讨论
2.1 重组表达载体 pMD-AOX-GOD的构建及阳性转化子的筛选
2.1.1 重组表达载体 pMD-AOX-GOD的构建
按照1.2.1方法构建重组表达载体pMD-AOX-GOD,目的基因PCR和酶切验证结果如图2所示,目的基因PCR得到大小约 1 700 bp 的目的片段,用XhoⅠ和NotⅠ双酶切质粒后得到1 700 bp和5 900 bp两个片段,均与理论值相符,重组表达载体构建成功。

图2 葡萄糖氧化酶的·PCR·扩增和重组质粒 pMD-AOX-GOD酶切鉴定
2.1.2 阳性转化子的筛选
一般情况下,整合到毕赤酵母基因组中的拷贝数越多重组菌株的抗性浓度会越高,这样可以通过不同抗性浓度的平板来筛选不同拷贝数的重组菌株[15]。利用不同 G418 浓度对重组质粒 pMD-AOX-GOD在毕赤酵母X33感受态中进行转化子的筛选,通过不同抗性浓度以期得到不同拷贝数的重组菌株。筛选平板上G418的浓度分别为 0.10、0.20、0.50、1.00、2.00 mg/mL,2-3 d 后1.00 mg/mL浓度以下的长出数量不一的单菌落,2.00 mg/mL G418 条件下3 d后开始生长零星菌落,并且菌落总数随着抗性浓度增加而减少,菌落也越来越小。望松柏等人[16]研究发现卡氏德巴利酵母植酸酶重组菌株在 G418 浓度为 2.00 mg/mL 条件下无法生长。Aw等人[17]的研究也发现高浓度 G418 会影响单菌落生长,这往往是初始抗性压力过大造成的。
挑取部分重组菌株进行试管水平发酵,甲醇诱导72 h后测定发酵上清液的GOD酶活(图3),重复3次结果显示在G418浓度为1.00 mg/mL的平板上筛选出一株GOD酶活高且稳定性好的菌株。

图3 筛选重组菌株发酵上清液的酶活检测
利用荧光定量PCR方法[18]测定该重组菌株中GOD基因拷贝数为4,高密度甲醇发酵诱导 120 h 后GOD酶活力达 426 U/mL,该菌株作为第一代高产量GOD重组菌,命名为 X33/AnGOD,制备成感受态备用。
2.2 基因多拷贝表达载体的构建及阳性转化子的筛选
2.2.1 多拷贝表达载体的构建
多表达盒载体的构建参照方法1.2.2,利用同尾酶BamHⅠ和BglⅡ 对pPICZαA-GOD进行双酶切,回收一段含有醇氧化酶基因启动子( 5′AOX1)、α-factor 信号因子、目的基因(god)和醇氧化酶基因终止子(3′AOX1) 的顺式作用元件,同时,用BamHⅠ对pPICZαA-GOD进行单酶切,将获得的线性片段与表达盒片段连接转化,筛选阳性克隆,通过双酶切对含有2拷贝表达盒的重组表达载体pPICZαA-GOD2进行验证(图4(a))。利用BamHⅠ/BglⅡ对pPICZαA-GOD2双酶切,获得2拷贝表达盒片段,将其与用BamHⅠ酶切线性化的pPICZαA-GOD2连接,获得含有4拷贝表达盒的重组表达载体 pPICZαA-GOD4,用同样的方法对其验证。使用同样的方法改造、构建和验证含有3拷贝、5拷贝表达盒的重组表达载体pPICZαA-GOD3、pPICZαA-GOD5。如图4(b)所示,用BamHⅠ/BglⅡ 对pPICZαA-GODn(n=1-5)双酶切后,分别在位置3.4 kb、6.8 kb、10.0 kb、13.6 kb、17.0 kb有5个片段,与理论值相符。结果表明已成功构建多拷贝表达盒表达载体。

图4 多拷贝表达载体的酶切检测
2.2.2 阳性转化子的筛选
将含有多拷贝表达盒的重组表达载体pPICZαA-GODn(n=1-5)分别用BamHⅠ 酶切线性化,分别电击转化后涂布于Zeocin抗性平板上,筛选含多拷贝基因的转化子。每个基因剂量选取部分菌株进行测定,其拷贝数目分别为1~5。挑取部分转化子测定试管发酵水平的酶活,用 SDS-PAGE 蛋白电泳检测分析发酵上清液中的蛋白。结果如图5所示,随着基因拷贝表达盒数量的增加,GOD 酶活力逐渐增加,4拷贝转化子酶活性最高,约为1拷贝重组菌株的3倍,但是,5拷贝GOD酶活反而下降了,说明在一定范围内,基因拷贝数的增加可以提高酶活水平。4拷贝基因重组菌株高密度甲醇发酵诱导 120 h 后GOD酶活力达 373 U/mL。
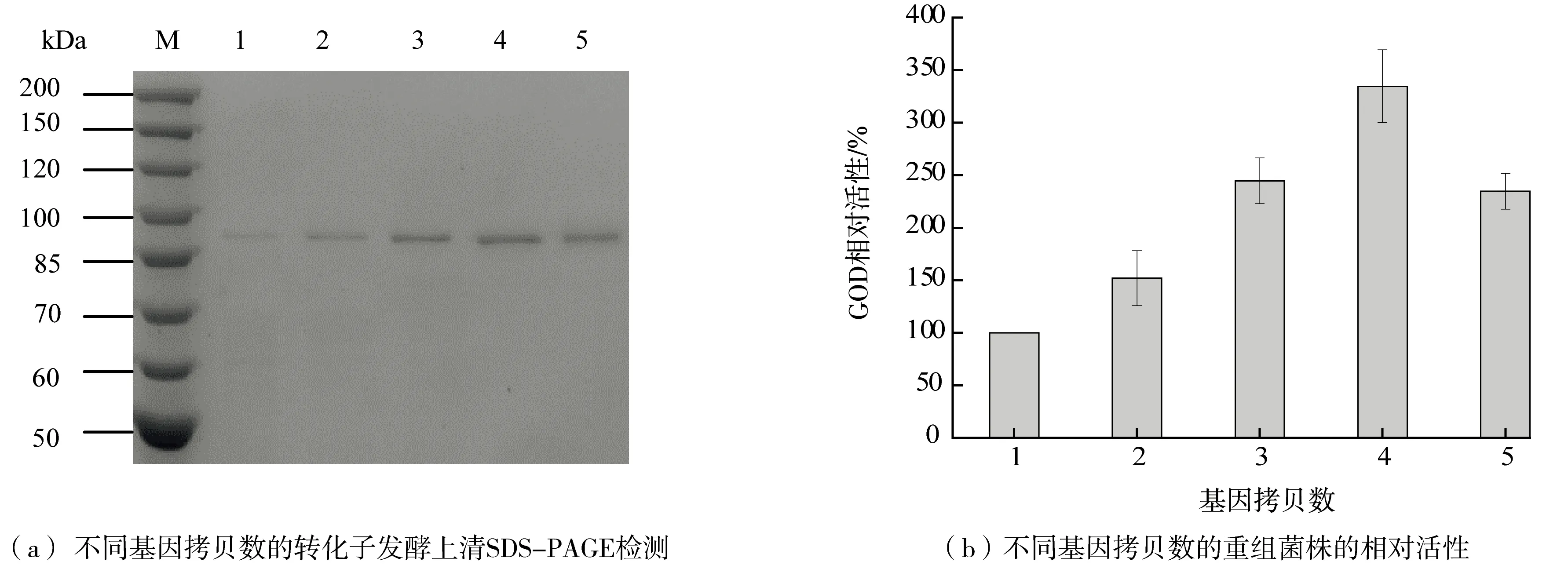
图5 拷贝数对蛋白表达水平的影响
结果表明,随着基因表达盒数量的增加,GOD 酶活逐渐增加,4拷贝转化子酶活性最高,但5拷贝GOD酶活出现下降,甚至低于3拷贝。这说明一定数量的基因拷贝数会增加外源蛋白的表达量,但拷贝数过多会影响外源蛋白的表达水平。Jiao等人[19]研究脂肪酶在毕赤酵母中高效表达时发现,基因拷贝数多于5个后脂肪酶的表达水平出现降低。陈姗姗[20]通过构建多拷贝串联hLYZ基因转化重组子研究人源溶菌酶在毕赤酵母中的高效表达,发现多拷贝重组子的溶菌酶表达性能较低。Wu等人[21]研究在毕赤酵母中高水平分泌HSA/GH融合蛋白时构建了1~7拷贝的重组菌株,发现含2~3个HSA/GH基因拷贝的转化子蛋白表达量显著增加,与7拷贝无显著差异。这是由于随着基因拷贝数的增加,mRNA翻译,蛋白质折叠可能受到了限制,大量未折叠或者错误折叠的蛋白积累,从而产生负反馈作用[22,23],因此,蛋白折叠分泌的研究也十分重要。
2.3 多拷贝分子伴侣共表达重组菌株的筛选及鉴定
将多拷贝HAC1、PDI表达载体分别用BamHⅠ酶切线性化,电击转化至X33/AnGOD 感受态后涂布于含G418和Zeocin 的双抗筛选平板上得到转化子。挑取阳性转化子进行试管水平诱导发酵,用1% 甲醇诱导3 d,发酵上清液测定GOD酶活力。结果如图6 所示,不同拷贝数的分子伴侣共表达重组菌株的GOD酶活,随着分子伴侣拷贝数的增加呈现出上升的趋势,但是超过一定的拷贝数后GOD酶活反而下降。其中,2拷贝的HAC1共表达重组菌株GOD酶活最高,比出发菌株提高67% ;1 拷贝的PDI共表达重组菌株GOD酶活最高,比出发菌株提高128%,将该菌株作为第二代高产量GOD重组菌,命名为X33/AnGOD-PDI。实验结果表明适当增加分子伴侣的拷贝数可以提高酶活。利用RT-qPCR 检测方法测定共表达重组菌株中分子伴侣HAC1、PDI的基因拷贝数,结果显示共表达重组菌株中分子伴侣HAC1、PDI的基因拷贝数符合预期。

图6 共表达分子伴侣对GOD表达水平的影响
很多参与蛋白折叠的分子伴侣已经被验证可以提高外源蛋白在毕赤酵母中的表达水平,如蛋白质二硫键异构酶 PDI[24-26]、内质网氧化还原酶 Ero1[24,27]、UPR 调节因子HAC1[6,28]等。通过整合分子伴侣蛋白以期提高GOD表达,结果显示与前人研究成果一致,整合分子伴侣的重组菌株 GOD酶活均比出发菌株高。
2.4 10 L发酵罐高密度发酵试验
将第一代菌株 X33/AnGOD 和整合分子伴侣PDI 的第二代菌株 X33/AnGOD-PDI 按上述条件进行10 L 发酵罐高密度放大培养。发酵上清液检测GOD酶活(图7),整合分子伴侣的菌株GOD酶活较第一代菌株有很大提高,第一代重组菌株在发酵结束时酶活为537 U/mL(图7(a)),而整合分子伴侣PDI的重组菌株发酵诱导 156 h 后酶活达到 1 198 U/mL,提高123%(图7(b))。
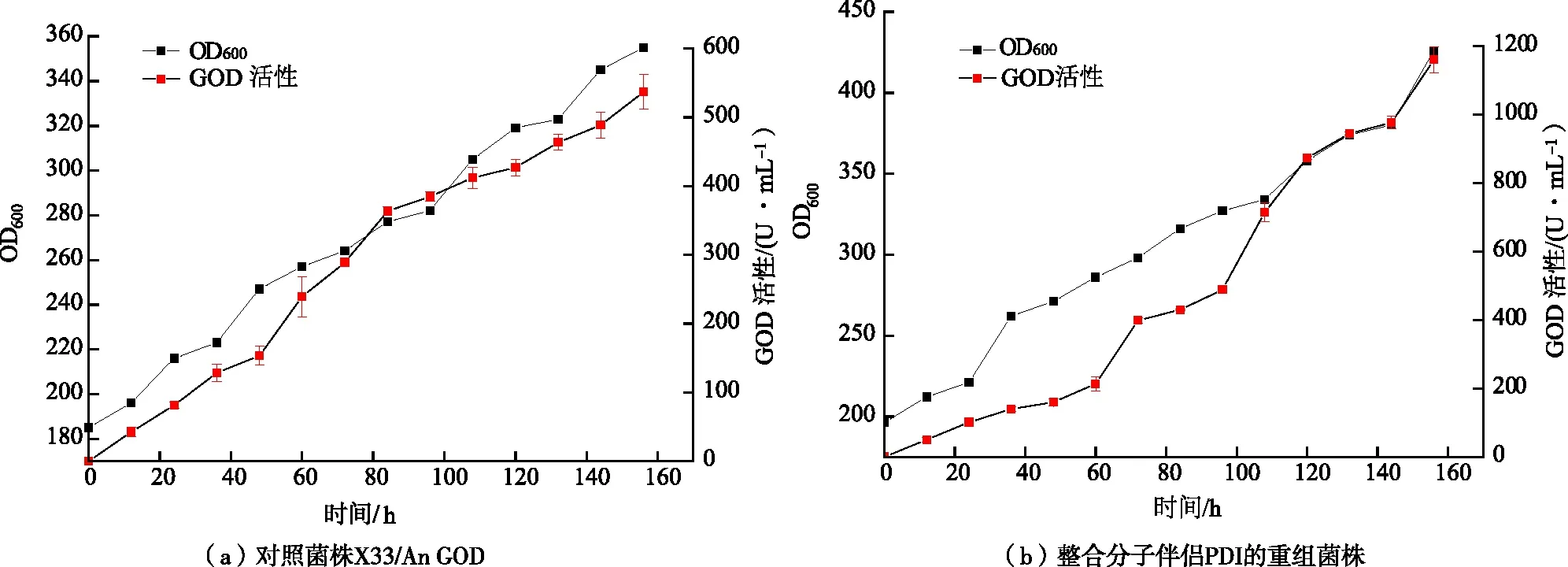
图7 在10 L发酵罐中的生长曲线和酶活力分析
3 结论
本研究利用抗生素浓度梯度、体外多拷贝构建筛选获得了第一代高产GOD重组菌株,以此为基础,本研究首次构建了分子伴侣HAC1、PDI多拷贝共表达重组菌株,1 拷贝的PDI共表达重组菌株X33/AnGOD-PDI中 GOD 表达量比出发菌株提高128%。通过10 L 发酵罐高密度发酵,酶活达到1 198 U/mL,与出发菌株相比,其发酵生产 GOD 的能力显著增加,表达量提高123%。使用的发酵培养基为BSM培养基,提高重组菌株中GOD酶活的同时大大降低了发酵成本,适宜大规模工业生产。
猜你喜欢
河北医学(2021年10期)2021-10-27 00:37:14
昆明医科大学学报(2020年12期)2021-01-26 00:44:02
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
中国生殖健康(2018年1期)2018-11-06 07:14:38
山东医药(2017年20期)2017-07-01 19:52:07
中国学术期刊文摘(2016年1期)2016-02-13 14:05:23
山东医药(2015年44期)2015-02-28 14:29:02
发明与创新(2015年25期)2015-02-27 10:39:16
医学综述(2014年24期)2014-03-08 07:07:24
计算机应用文摘(2010年30期)2010-04-29 00:44:03
