
患呼吸道疾病羊鼻腔及生活环境微生物多样性分析
2023-08-25 09:03郝秀静
畜牧兽医学报 2023年8期
张 颖,金 华,韩 杨,眭 丹,肖 鑫,郝秀静*,李 敏*
(1.宁夏大学西部特色生物资源保护与利用教育部重点实验室,银川 750021;2.宁夏大学生命科学学院,银川 750021;3.宁夏中牧亿林畜产股份有限公司,银川 750021;4.宁夏农业勘察设计院,银川 750021)
呼吸系统疾病作为一种高发病,严重影响着动物健康[1-3]。羊呼吸系统疾病是冬春季节羊养殖业面临的棘手问题之一,其导致感染羊群健康状况不佳,生长缓慢,发病率和死亡率增加以及治疗和疫苗接种成本增加,给养羊业带来了严重的经济损失[4-5]。羊呼吸道疾病的发生和发展一方面与致病性细菌、病毒和支原体等的感染密切相关,其中,多杀性巴氏杆菌(Pasteurellamultocida)[6]、溶血性曼氏杆菌(Mannheimiahaemolytica)[7]、山羊副流感病毒3型(Caprineparainfluenzavirus type 3)[8]和绵羊肺炎支原体(Mycoplasmaovipneumoniae)[9]等较为常见。另一方面,呼吸道中定植的优势益生菌群在抵抗病原菌感染中发挥重要作用,Timsit等[10]研究发现,健康公牛中乳酸乳球菌和干酪乳杆菌能够抵抗细菌性病原体在呼吸道中的定植。最近的研究表明,呼吸道中多种病原体的协同感染会改变呼吸道中细菌的群落结构,进而对机体造成更大的损害[11]。Zhang等[12]发现呼吸道中具有致病潜力的常驻细菌与流感病毒的合并感染,通过影响微生物群落结构和整体微生物基因表达,间接影响呼吸道中抗生素耐药基因的表达,从而使疾病恶化。Shobo等[13]研究发现,环境是细菌病原体的“蓄水池”,可能是疾病发生发展的潜在风险因素。环境因素,如灰尘、气溶胶和湿度等在维持呼吸系统微生物稳态方面和调节病原体致病性方面也扮演着重要的角色,其不仅能够降低宿主的防御机制,还能够增加宿主的易感性[14-15]。近年来,随着高通量测序技术的发展,16S rRNA基因测序有助于全面识别与疾病相关的微生物菌群变化[16]。鼻腔作为呼吸道的起始部位,是阻挡病原体入侵机体的第一道防线[17]。鼻咽中微生物群的群落结构和丰度可能影响整个呼吸道的健康,对于防止呼吸道病原体定植具有积极意义[18],但有关羊鼻腔微生物菌群的多样性及其与疾病的关系鲜有报道,而且其群落结构是否受其生活环境影响尚不清楚。因此,本研究采用16S rRNA测序技术,获得了羊鼻腔及羊舍地面表层堆积物的微生物菌群,以健康羊和其所在羊舍地面表层堆积物为对照,分析患呼吸道疾病羊鼻腔及其所在羊舍地面表层堆积物的群落结构、alpha多样性及beta多样性,旨在探究患呼吸道疾病羊鼻腔及其生活环境中微生物菌群的变化,分析羊患呼吸道疾病与其生活环境的菌群结构之间的联系,为进一步开展羊呼吸系统微生物多样性研究提供数据基础,同时为羊呼吸系统疾病的预防和治疗提供理论依据。
1 材料与方法
1.1 样本采集
2019年4月,从宁夏回族自治区银川市某一规模化羊养殖场采集3~4月龄羊鼻腔拭子和羊舍地面表层堆积物(粪便、垫料,毛发和尘土的混合物)。在收集样本之前对羊只进行跟踪调查和健康检查,记录具有咳嗽、喘息、流鼻涕和消瘦等典型患呼吸道疾病症状的羊。使用无菌一次性棉棒采集10份患呼吸道疾病羊的鼻腔拭子样本(每份鼻拭子收集4个重复样本),设为D组,采集6份健康羊鼻腔拭子作为对照,设为H组。同时,在每一羊舍地面表层堆积物区域随机选取4处位置,按照10 cm的深度采集样本,将4处样本混合后作为一份样本进行后续试验。共采集8份患呼吸道疾病羊所在羊舍地面表层堆积物样本,设为ED组,采集7份健康羊所在羊舍地面表层堆积物样本,设为EH组。所有样本均被密封,详细登记其分组及编号信息,干冰保存运送至北京诺禾致源科技股份有限公司进行后续测序。
1.2 羊鼻腔拭子和羊舍地面表层堆积物微生物测定
使用QIAamp DNA Mini Kit (QIAGEN公司,Germany)和基因组DNA 提取试剂盒(天根生物科技有限公司)分别提取羊鼻腔拭子和羊舍地面表层堆积物样本中的总DNA,利用NanoDrop-8000检测样本DNA浓度和纯度,并用1%琼脂糖凝胶电泳检测DNA的完整性。选择细菌 16S rRNA V3-V4 区域进行 PCR 扩增,并利用QIAquick Gel Extraction Kit(QIAGEN公司,Germany)纯化 PCR 产物。基于IonS5TMXL测序平台,构建小片段文库进行单端测序。
1.3 16S rRNA数据处理
测序完成后,用Cutadapt(Martin M.2011)软件对原始数据进行过滤和质控,主要工作包括剪切末端低质量较多的部分、拆分样本数据、去除reads中的接头序列和barcode序列并过滤掉序列中的嵌合体序列。质控得到有效数据后,以97%的一致性将序列聚类成为操作分类单元(operational taxonomic units,OTUs),然后与Silva132数据库比对,对OTUs序列进行物种注释。
1.4 微生物结构组成和多样性分析
基于样本的 OTUs 聚类和注释,样本测序深度、物种的丰富度和均匀度由稀释曲线和等级聚类曲线表示。微生物菌群alpha多样性由观测物种数、Simpson指数、Shannon 指数、Chao 1指数和ACE指数表示。计算各组样本在不同分类单元的相对丰度,并分别在门、纲、目、科、属和种水平上构建矩阵。基于Unweighted-UniFrac距离的主坐标分析(principal coordinate analysis,PCoA)和多响应排列程序分析(multiple response permutation procedure,MRPP)用于显示微生物群落的beta多样性。最后,效应大小的线性判别分析(linear discriminant analysis of effect size,LEfSe)用于估算每个物种丰度对差异效果影响的大小,从而在组与组之间寻找具有统计学差异的生物标识(biomarker),即组间差异显著的物种(logLDA分数>4被认为具有显著差异)。
1.5 数据分析
采用GraphPad Prism 7 软件对alpha多样性指数及相关数据进行独立样本t检验或秩和检验,结果用“平均值±标准误(standard error,SE)”表示,P>0.05被认为没有统计学意义,P<0.05 被认为具有统计学意义(P<0.05为差异显著性,P<0.01和P<0.001为差异极显著)。
2 结 果
2.1 16S RNA测序结果及质控
基于IonS5TMXL测序平台,对羊鼻腔拭子和羊舍地面表层堆积物样本进行单端测序后,每个样本平均测得77 810条原始序列。对原始数据进行拼接和过滤后,如表1所示,每个样本平均得到73 215条有效数据,平均长度为408.61 nt,Q20占比80.24%,GC含量为52.12%,质控有效率达94.08%。

表1 数据质控平均数统计结果
以97%的一致性将有效序列聚类成为OTUs,分析样本测序深度和样本中物种的丰富度和均匀度。由图1A可知,所有样本的稀释曲线随着测序深度的增加逐渐趋于平缓,表明当前测序深度足够反映该群落样本所包含的微生物多样性。由等级聚类曲线(图1B)可知,所有样本在横轴上的跨度较大,而在纵轴上趋于平缓,表明样品中物种的丰富度和均匀度良好。
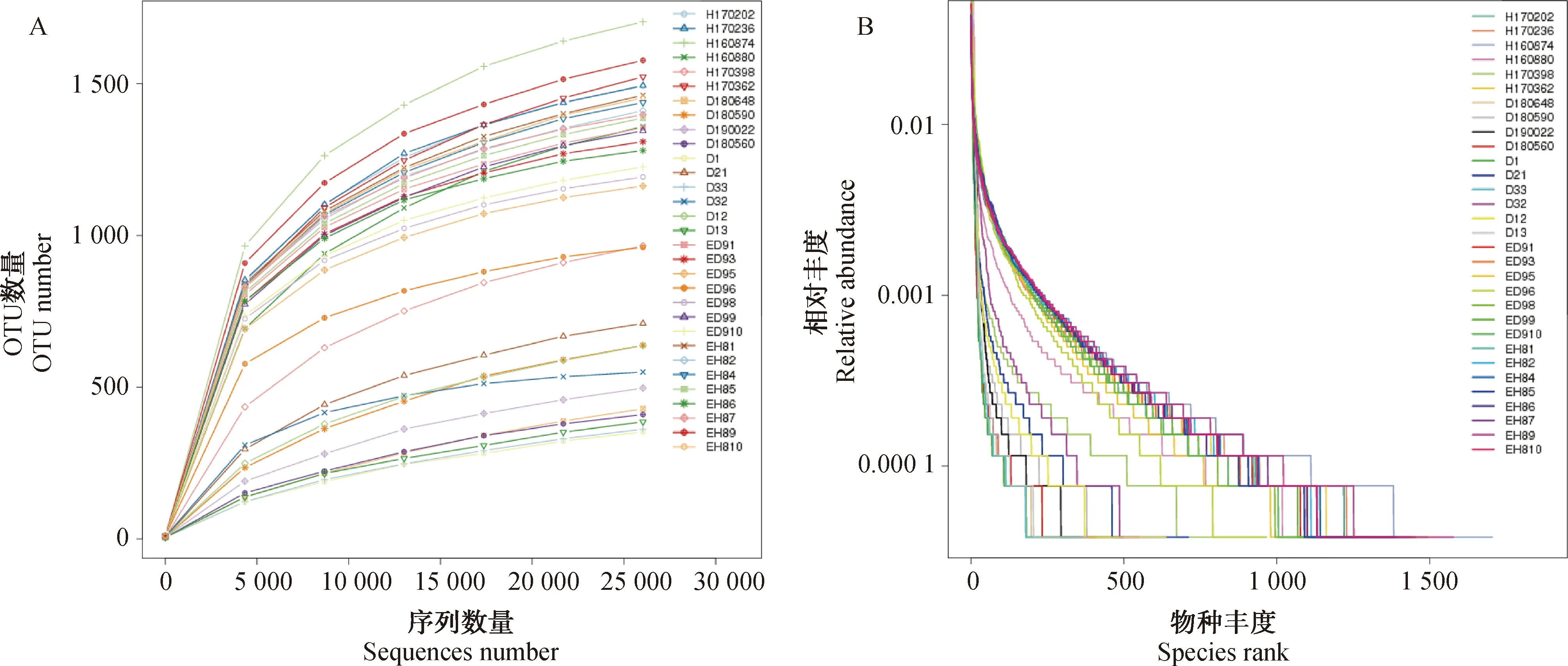
图1 所有样本稀释曲线和等级聚类曲线
2.2 羊鼻腔及周边环境中微生物菌群OTUs聚类
本试验共得到2 918个OTUs,如图2A所示,健康羊(H组)和患呼吸道疾病羊(D组)中共有的OTUs数为1 389个,H组中特有的OTUs数明显多于D组。如图2B所示,健康羊所在羊舍地面表层堆积物(EH组)中特有的OTUs数多于患呼吸道疾病羊所在羊舍地面表层堆积物(ED组),EH组和ED组共有的OTUs数为1 804个。H组和EH组中共有的OTUs数为1 907个(图2C)。ED组中特有的OTUs数多于D组,二者共有的OTUs数为1 234个(图2D)。上述结果表明,不同组别之间菌种的丰度具有差异,健康羊鼻腔及其所在羊舍地面表层堆积物中所含菌种的丰度较高,而患呼吸道疾病羊鼻腔及其所在羊舍地面表层堆积物中所含菌种丰度较低。
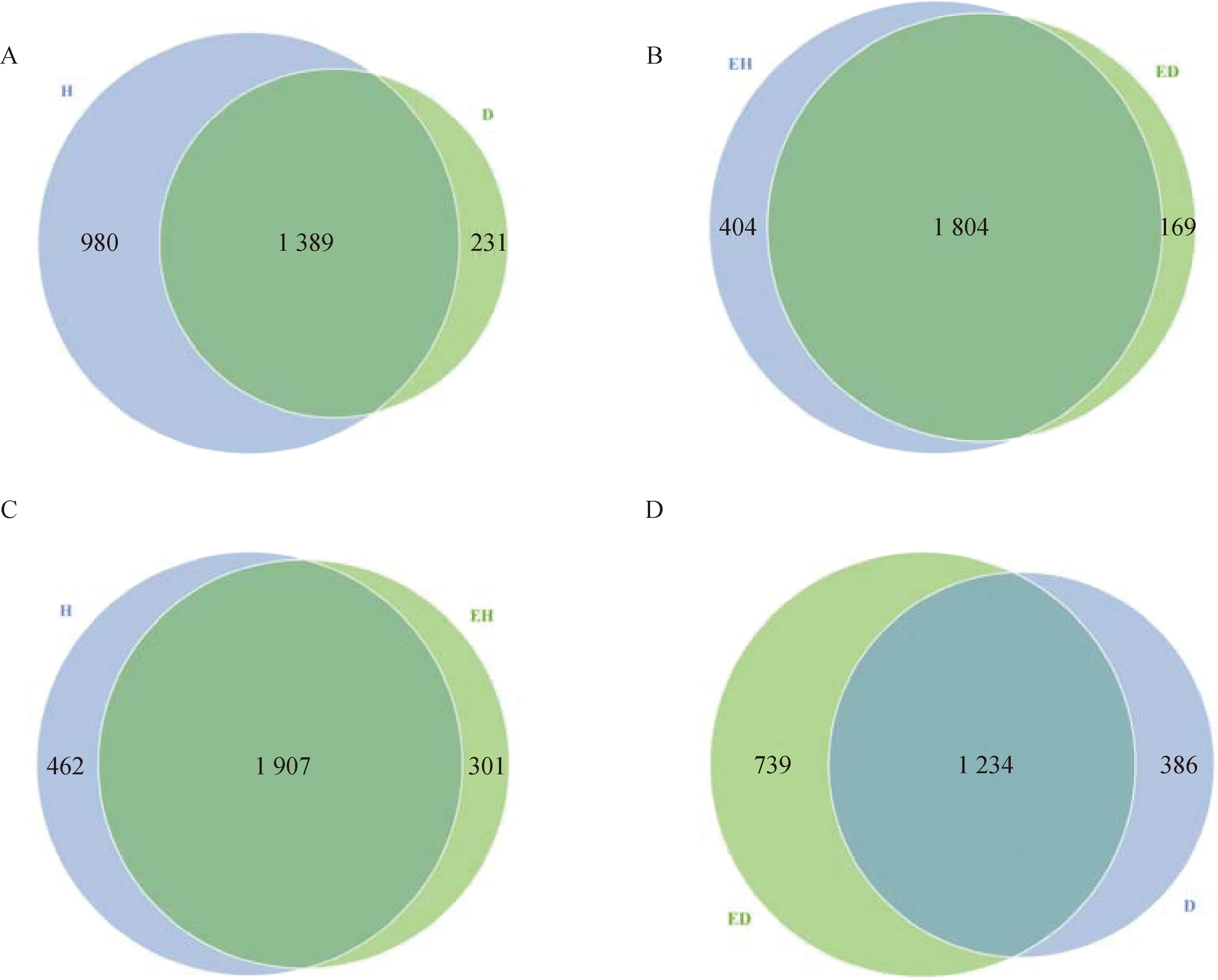
图2 羊鼻腔及羊舍地面表层堆积物中微生物菌群OTUs聚类
2.3 羊鼻腔及周边环境中微生物alpha多样性分析
为了进一步分析羊鼻腔及其生活环境中微生物群落的丰富度和多样性,对测序数据进行alpha多样性分析。各组样本的覆盖度指数均为0.9~1.0,表明测序结果能反映各组菌群多样性组成(图3A)。与健康羊(H组)相比,患呼吸道疾病羊(D组)中观测物种数、Simpson指数、Shannon 指数、Chao 1指数和ACE指数均显著下降(P<0.01)(图3B~F),表明患呼吸道疾病羊鼻腔微生物的多样性和丰富度降低。进一步比较健康羊所在羊舍地面表层堆积物(EH组)和患呼吸道疾病羊所在羊舍地面表层堆积物(ED组)中Chao 1指数和ACE指数,发现ED组中Chao 1指数和ACE指数均显著低于EH组(P<0.05)(图3E~F),表明患呼吸道疾病羊生活环境中菌群丰富度也降低。
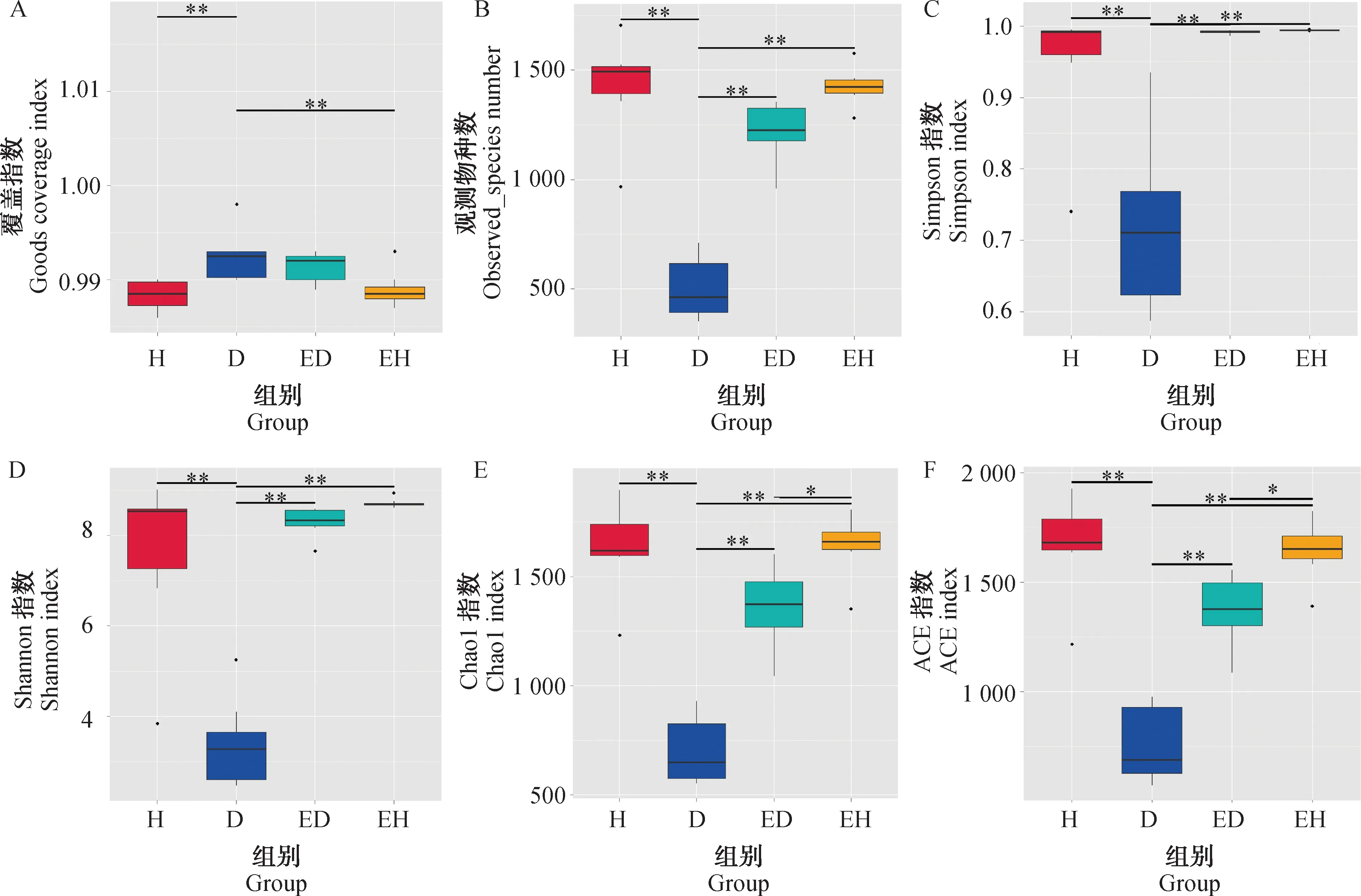
*.P<0.05;**.P<0.01
2.4 羊鼻腔及周边环境中微生物群落结构
本试验共鉴定出个32个门(Phylum),51个纲(Class),97个目(Order),175个科(Family),359个属(Genus),237个种(Species)。各组中占据主导地位的菌门主要包括变形菌门(Proteobacteria)(H组:26.42%;D组:79.99%;ED组:3.05%;EH组:5.11%)、厚壁菌门(Firmicutes)(H组:53.01%;D组:6.08%;ED组:63.04%;EH组:65.14%)和拟杆菌门(Bacteroidetes)(H组:11.93%;D组:9.05%;ED组:23.92%;EH组:21.84%)。与H组相比,D组中变形菌门的相对丰度增加,而厚壁菌门相对丰度降低。与EH组相比,ED组中变形菌门和厚壁菌门的相对丰度增加,而拟杆菌门的相对丰度降低(图4A)。
在纲水平上(图4B),各组占据主导的菌群均为变形菌纲(Gammaproteobacteria)、梭菌纲(Clostridia)和拟杆菌纲(Bacteroidia),与H组相比,D组鼻腔中变形菌纲相对丰度增加(H组:24.98%;D组:79.41%),梭菌纲相对丰度降低(H组:43.98%;D组:4.43%)。假单胞菌目(Pseudomonadales)(H组:1.38%;D组:66.30%;ED组:0.41%;EH组:0.47%)、黄色单胞菌目(Xanthomonadales)(H组:18.81%;D组:5.51%;ED组:0.23%;EH组:1.98%)和梭菌目(Clostridiales)(H组:43.97%;D组:4.43%;ED组:60.48%;EH组:62.14%)均是各组目水平上的优势物种(图4C),它们在EH组和ED组的相对丰度无明显变化,H组中黄色单胞菌目和梭菌目相对丰度较高,而D组中黄色单胞菌目和梭菌目的丰度明显降低,假单胞菌目丰度明显增加。
在科水平上(图4D),黄色单胞菌科(Xanthomonadaceae)(H组:18.81%;D组:5.49%)和瘤胃菌科(Ruminococcaceae)(H组:23.17%;D组:2.48%)是H组和D组的优势物种,但莫拉菌科(Moraxellaceae)在两组中相对丰度差异较为明显,其在D组中的相对丰度较高(H组:1.30%;D组:66%)。在属水平上(图4E),与H组相比,D组中莫拉菌属(Moraxella)(H组:1.22%;D组:66.23%)、伯杰菌属(Bergeyella)(H组:0.04%;D组:3.68%)和丝状杆菌属(Filobacterium)(H组:0.26%;D组:2.33%)的相对丰度明显增加,而寡养单胞菌属(Stenotrophomonas)(H组:18.72%;D组:5.46%)的相对丰度明显降低。除此之外,支原体属(Mycoplasma)(H组:0.03%;D组:1.93%)、波氏杆菌属(Bordetella)(H组:0.00%;D组:1.79%)和曼氏杆菌属(Mannheimia)(H组:0.32%;D组:1.08%)等细菌性病原的相对丰度在D组中也有明显增加。通过与数据库Silva比对,注释到种水平的OTUs比例为9.53%,如图4F所示,在已注释到的种中,D组的优势种为腔隙莫拉菌(Moraxella_lacunata)(H组:0.03%;D组:1.63%)和动物溃疡伯杰菌(Bergeyella_zoohelcum)(H组:0.03%;D组:1.12%)。
上述结果提示,羊鼻腔及羊舍地面表层堆积物样品中微生物学分类具有多样性,各组样品的分类结构相似,但部分物种相对丰度差异较大。患呼吸道疾病羊鼻腔与健康羊鼻腔微生物的优势种属存在明显差异,患呼吸道疾病羊鼻腔中明显增加的种属,如:莫拉菌属(Moraxella)、伯杰菌属(Bergeyella)等相对丰度的增加可能是引起羊患病的主要病原微生物。
2.5 羊鼻腔及周边环境中微生物菌群beta多样性分析
为了更好地了解羊鼻腔及生活环境中微生物群的动态演替规律,基于Unweighted-UniFrac距离进行PCoA分析,如图5A所示,样本之间存在明显的聚类分离,表明各组具有不同的群落结构。由Beta多样性组间差异分析的箱型图可知(图5B),D组和H组的群落结构具有极显著差异(P<0.01),ED组和EH组的群落结构也具有显著差异(P<0.05)。另外,MRPP分析(表2)也得到了同样的结果。以上数据证实患呼吸道疾病羊的微生物群落组成发生了显著变化。
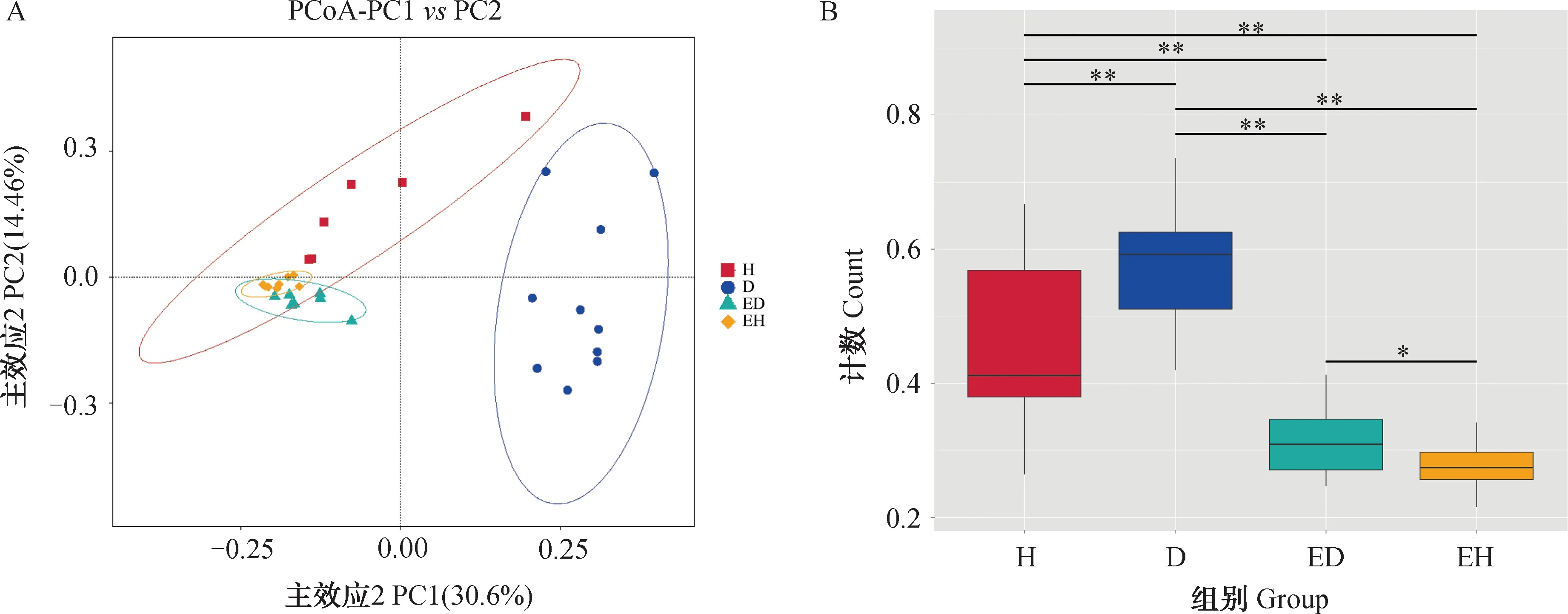
*.P<0.05;**.P<0.01
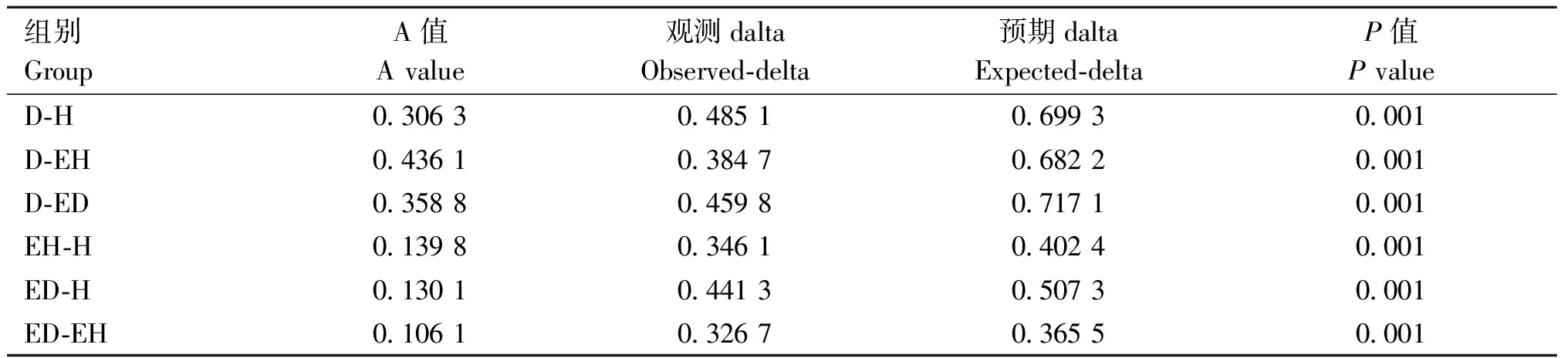
表2 MRPP 分析组间差异显著性
2.6 羊鼻腔及周边环境中差异微生物分析
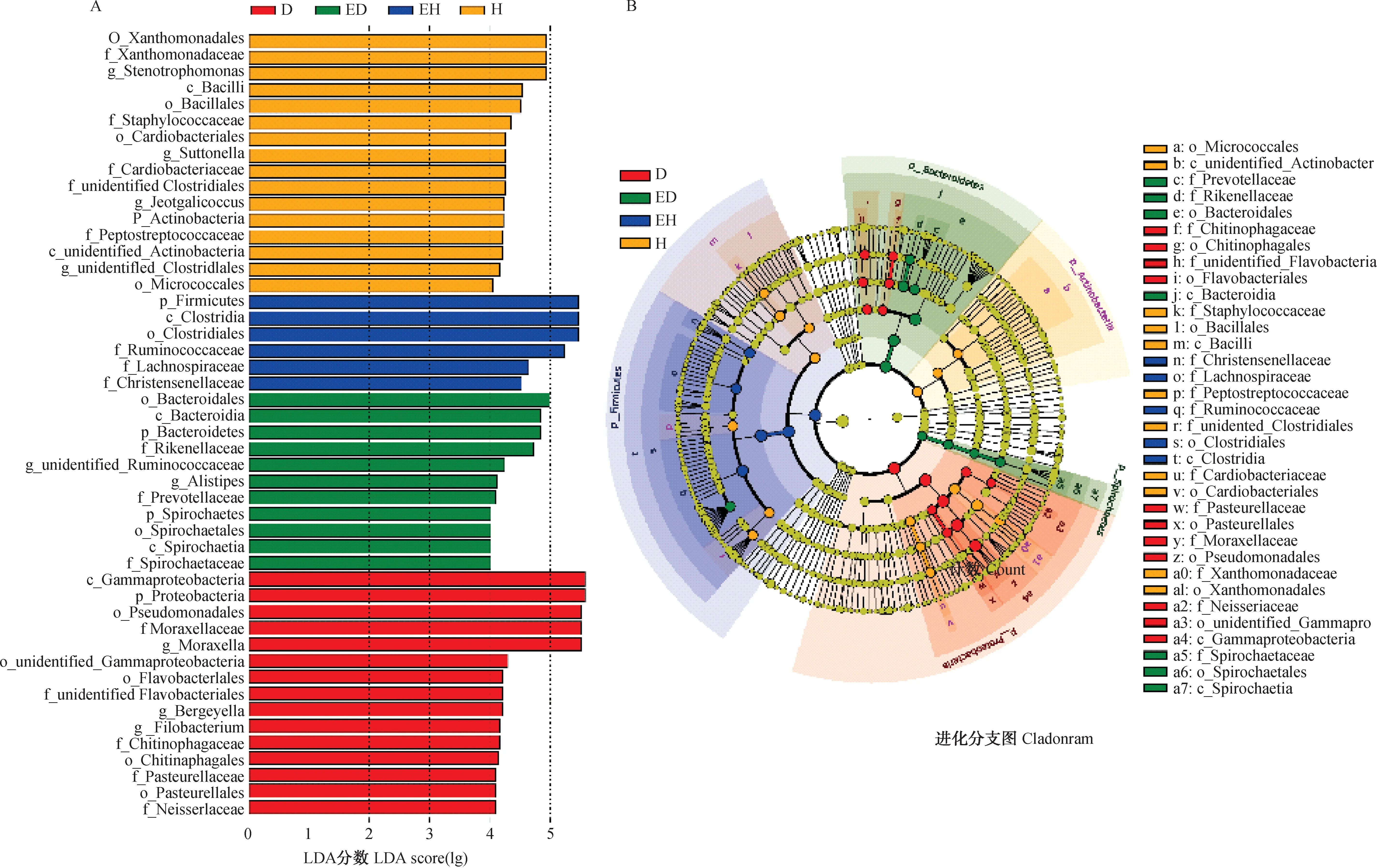
为了明确健康羊鼻腔及周边环境与患呼吸道疾病羊鼻腔及周边环境之间显著变化的微生物,作者利用LEfSe分析对差异微生物进行筛选,由图6可知,LAD>4的微生物标志物共有48个,厚壁菌门(Firmicutes)是EH组具有显著差异的微生物菌门,而拟杆菌门(Bacteroidetes)是ED组具有显著差异的微生物菌门。H组共获得16个差异微生物,D组共获得15个差异微生物。在属水平上,H组具有显著差异的微生物菌群为寡养单胞菌属(Stenotrophomonas),而莫拉菌属(Moraxella)、伯杰菌属(Bergeyella)和丝状杆菌属(Filobacterium)等是D组具有显著差异的微生物菌群。经分析,未发现H组和D组有共同的差异显著的微生物菌群,这可能与健康羊鼻腔和患呼吸道疾病羊鼻腔具有不同的微生物群落结构有关。
3 讨 论
传统的微生物分离培养技术难以分离不易培养的、微量的微生物,不利于全面识别与疾病相关的微生物菌群变化。近年来,随着高通量测序技术的快速发展,16S rRNA基因测序技术逐渐成为了研究微生物群落组成和分布的重要手段[19-20]。鼻咽部作为呼吸道的起始部位,其内定植的微生物群是一个动态而复杂的生态系统,在抵御病原体感染,维持呼吸道健康中具有重要调控作用[21-22]。研究发现,呼吸系统生态失衡与呼吸道疾病的发生发展密切相关,其微生物群的组成和多样性的差异是导致疾病的重要因素[23-24]。
本研究通过16S rRNA基因组测序技术分析健康羊和患呼吸道疾病羊鼻腔及所在羊舍地面表层堆积物微生物菌群的结构。alpha多样性和beta多样性分析结果显示:健康羊鼻腔与患呼吸道疾病羊鼻腔具有不同的微生物群落结构,二者之间具有极显著的差异(P<0.01)。进一步分析发现,变形菌门是患呼吸道疾病羊鼻腔中显著增加的菌门。在属水平,造成健康羊鼻腔和患呼吸道疾病羊鼻腔菌群结构显著差异的属为寡养单胞菌属、莫拉菌属、伯杰菌属和丝状杆菌属。此前的研究表明,寡养单胞菌属是一类革兰阴性菌,至少包括8个种,其存在于整个环境中,土壤和植物来源的寡养单胞菌属有利于生物修复和植物健康[25],而嗜麦芽寡养单胞菌作为寡养单胞菌属唯一一类被报道的致病菌,其在动物中是一种机会性致病菌[26]。莫拉菌属是牛和猪上呼吸道中最丰富的属之一,其可单独或与其他致病菌协同感染引起疾病[27-28]。最近,董文龙等[29]、Li等[30]相继从患有呼吸道疾病的羊鼻腔中分离出了莫拉菌,可见其在羊呼吸道疾病中具有重要的潜在致病性。Lorenzo等[31]从仔猪鼻道中分离出的伯杰菌属在体外表现出明显的潜在毒力特征,如:抵抗吞噬、耐受血清和黏附上皮细胞等。丝状杆菌属被鉴定为啮齿类动物的主要病原菌[32]。综上所述,患呼吸道疾病羊鼻腔中显著增加的莫拉菌属、伯杰菌属和丝状杆菌属等可能是引起羊患呼吸道疾病的重要的潜在致病因子。
越来越多的研究表明,环境中的微生物群对疾病发展具有潜在的影响[33-34]。Volenis-Chacin等[35]对猪气管微生物群与空气、粪便和口腔液中微生物群的相关性分析发现来自不同生态位的细菌群落之间存在复杂的相互作用。本研究中,作者分析了羊生活环境中微生物多样性与羊呼吸道疾病之间的关系,发现健康羊所在羊舍地面表层堆积物与患呼吸道疾病羊所在羊舍地面表层堆积物的菌群结构具有显著差异(P<0.05)。厚壁菌门和拟杆菌门是健康羊所在羊舍地面表层堆积物和患呼吸道疾病羊所在羊舍地面表层堆积物中具有显著差异的微生物菌门,患呼吸道疾病羊所在羊舍地面表层堆积物中厚壁菌门的相对丰度高于健康羊所在羊舍地面表层堆积物。然而,患呼吸道疾病羊鼻腔显著变化的菌门为变形菌门。在属水平,寡养单胞菌属是健康羊鼻腔和患呼吸道疾病羊鼻腔中具有显著差异的属,其在健康羊鼻腔中的相对丰度高于患呼吸道疾病羊鼻腔,而其在健康羊所在羊舍地面表层堆积物和患呼吸道疾病羊所在羊舍地面表层堆积物中并无显著变化。在患呼吸道疾病羊鼻腔中相对丰度较高的莫拉菌属、伯杰菌属和丝状杆菌属在患呼吸道疾病羊所在羊舍地面表层堆积物中的相对丰度也无显著变化。由此可见,羊生活环境中微生物菌群结构的改变对羊呼吸道疾病的发生和发展无直接联系,但其与羊鼻腔微生物群之间复杂的相互作用及对羊呼吸道疾病的潜在危害需进一步探讨。
4 结 论
4.1患呼吸道疾病羊鼻腔及其生活环境中微生物的丰富度和均匀度低于健康羊。
4.2莫拉菌属、伯杰菌属和丝状杆菌属是患呼吸道疾病羊鼻腔中相对丰度较高的差异菌属,支原体属、波氏杆菌属和曼氏杆菌属等细菌性病原相对丰度的增加以及患呼吸道疾病羊鼻腔中菌群结构的改变,可能是引起羊患呼吸道疾病的重要因素。
4.3本研究未发现羊生活环境中微生物菌群结构与羊呼吸道疾病的发生和发展的直接联系,但其潜在的风险仍需进一步研究。
猜你喜欢
国外核新闻(2022年9期)2022-12-16
青海畜牧兽医杂志(2021年6期)2022-01-24
中国比较医学杂志(2020年4期)2020-05-26
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
世界建筑导报(2018年4期)2018-09-21
西南石油大学学报(自然科学版)(2018年2期)2018-06-26
现代农村科技(2016年11期)2016-07-25
中国畜牧兽医文摘(2015年10期)2015-03-02
