
过渡金属磷化物的改性方法及其在电化学析氢中的应用
2023-08-16 09:06王蕴青杨国锐延卫
化工进展 2023年7期
王蕴青,杨国锐,延卫
(1 西安市固体废物资源再生与循环利用重点实验室,陕西 西安 710049;2 西安交通大学环境工程系,陕西 西安710049;3 西安交通大学应用化学系,陕西 西安 710049)
氢能作为具有高质量能量密度且环保的二次能源,展现出了能够替代传统化石能源的巨大潜力。但地球上没有天然储备的氢能源,需要通过制氢手段获取后才能对其进行利用[1]。利用风能或太阳能等可再生能源提供电能来电解水制氢已经成为最有前景的绿氢制取途径[2-4]。
在电解水制氢过程中,电极表面的催化剂材料是影响电化学析氢(hydrogen evolution reaction,HER)的重要因素,开发廉价、高效、稳定的电解水催化剂能够降低反应的过电位,有效提高析氢效率[5-6]。目前贵金属Pt 是性能最佳的HER 催化剂,但其储量稀缺、价格高昂,无法实现大规模的制氢应用,因此需要研究来源丰富、低廉高效的非贵金属催化剂来代替Pt 族催化剂。过渡金属磷化物(transition metal phosphides,TMPs)与氢化酶的结构和性质相似,具有稳定的催化活性和能够作为析氢、析氧催化剂的双功能特性,被认为是具有广阔应用前景的HER催化剂材料[7]。
TMPs由磷(P)和过渡金属(M)组成。由于其特殊的结构与性质,TMPs 具有十分多样的M/P化学计量比。根据M/P 原子比可以将TMPs 分为富磷磷化物(M/P<1)、单磷磷化物(M/P=l)和富金属磷化物(M/P>l)[8]。其中,单磷磷化物和富金属磷化物的性质类似于金属,具有良好的导电性。
几乎所有的过渡金属元素都可以被磷化形成TMPs。根据金属元素种类不同,过渡金属磷化物可以分为两大类:ⅥB族TMPs(如MoP和WP)和Ⅷ族TMPs(如NiP和CoP)[8-9]。但研究发现,只有6 种过渡金属(Fe、Co、Ni、Cu、Mo 和W)的磷化物在HER中展现出了较高的催化活性和稳定性,其中,镍磷化物由于与氢化酶的催化机理最为相似,得到了更为广泛的研究。此外,研究较多的还包括有Co与Fe的磷化物。
M/P 化学计量比对TMPs 尤为重要,可以影响TMPs 的性质、结构和制备过程等(图1)。因此除了对TMPs 的制备条件、组成元素种类和结构等方面进行调控,还可以通过调整TMPs 的M/P 化学计量比来优化其HER性能。

图1 TMPs作为HER电催化剂的主要研究方向
1 TMPs的主要性质
磷原子由于其半径较大,无法进入金属原子的密堆八面体中,因此TMPs 中的磷原子会互相结合形成柔韧性较高的磷骨架,金属原子则分布在磷原子周围形成三棱柱结构。M与P结合时,会形成丰富的金属-金属键(M—M 键)、磷-磷键(P—P键)以及金属-磷键(M—P 键),这也使得金属磷化物具有多样的组成和结构[10-12]。TMPs的大致结构如图2所示。
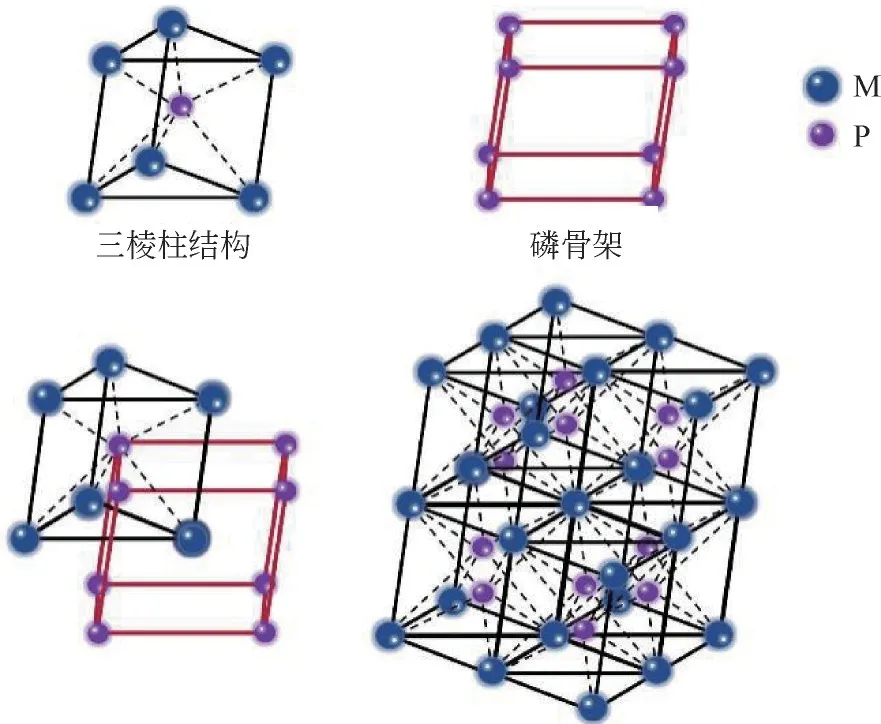
图2 TMPs的结构示意图
TMPs 的性质取决于M 的种类、M/P 化学计量比和晶体结构等[13]。在电化学析氢方面,由于其独特的电子结构,TMPs主要表现出良好的催化活性、导电性、耐腐蚀性以及电化学稳定性。
1.1 催化活性
HER 反应主要包括Volmer、Heyrovsky 和Tafel三个反应阶段。如图3所示,Volmer反应指电子转移得到吸附态氢的过程;Heyrovsky 反应指吸附在催化剂活性位点上的H与电解质中的质子结合的过程;Tafel 反应是指吸附在催化剂活性位点上的H相互结合的过程。一般析氢反应有Volmer-Heyrovsky 反应和Volmer-Tafel 反应两种过程[14-16]。根据电解质的不同,HER 的上述三个过程也不尽相同。在酸性介质中,首先是质子发生Volmer 反应,然后是Heyrovsky 或Tafel 反应。但碱性和中性介质中不存在自由质子,第一步要进行水分子的分解和OH-的解吸,这一过程导致了碱性或中性介质中的HER反应活性远远低于酸性介质[2,17]。
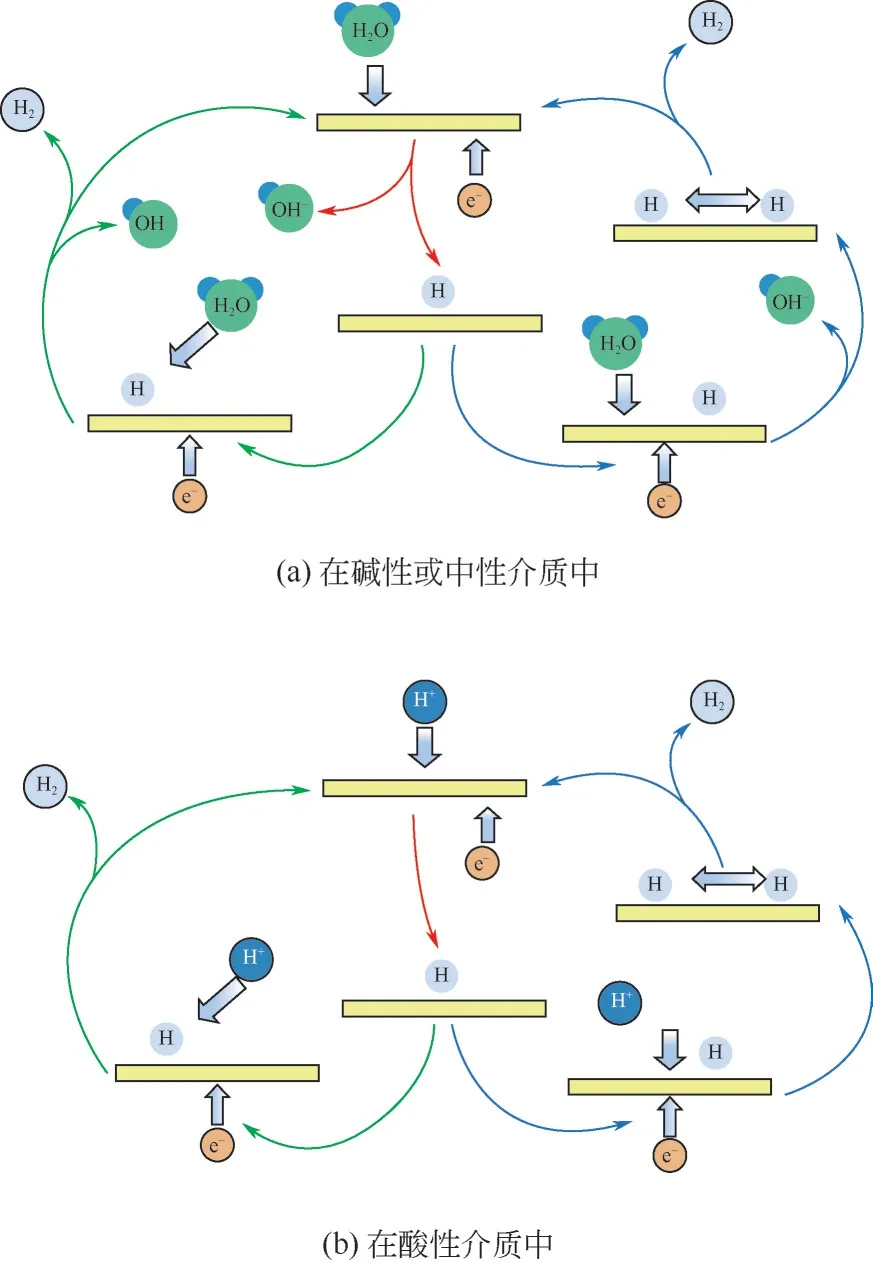
图3 电解水反应机理图 [18]
P 在TMPs 的电化学析氢过程中起着十分重要的作用。P 原子的引入使金属原子d 带收缩,导致费米能级附近的电子态密度增强,因此TMPs 具备了贵金属的特性,展现出极佳的催化性能[15]。在催化过程中,TMPs 中的磷和金属位点分别充当了质子受体位点和氢化物受体位点。P 原子带有负电荷,使得金属与H之间的化学键强度减弱,从而能够促进H的解吸。此外,P原子的引入还能使其与反应产物形成强度适中的化学键,减小纯金属对H2的强吸附,从而加快H2的脱离[11,19]。
相关研究发现,P 含量对TMPs 的HER 性能有着明显的影响,具有相同或相似结构、不同M/P比的TMPs 所表现出的HER 性能存在差异[20]。Schipper 等[21]发现富铁相磷化物的析氢活性明显优于富磷相,其具体的HER 活性表现为Fe3P>Fe2P>FeP。其他磷化物也展现出了相似的规律:在Ni2P、Ni5P4和Ni12P5中,P 含量最高的Ni5P4表现的HER活性最高;Co2P和CoP相比,CoP过电位明显低于Co2P[22];同样,MoP 的HER 活性也优于Mo3P[11]。由此可见,在一定范围内增加P原子含量可以有效地提高TMPs的HER活性。
铁族金属在高电流密度下具有良好的HER 性能。其中Ni、Co、Fe基磷化物已经被证明是TMPs中性能十分优异的电解水制氢催化剂。
(1)Ni 基磷化物的结构与性质与镍铁氢化酶极为相似,其中所含的化学键主要为Ni—P 键[23]。表1 总结了不同条件下合成的Ni 基磷化物的HER性能。

表1 常见Ni基磷化物的HER性能
(2)Co 基磷化物作为一种高活性和高稳定性的HER催化剂,主要包括CoP和Co2P两种相[14]。与前述规律相同,其分子中P含量越多,催化剂表面暴露出的活性位点就越多,这使得相同条件下的Co2P 的HER 性能优于CoP[22,38]。表2 对不同条件下合成的Co基磷化物的HER性能进行了总结。
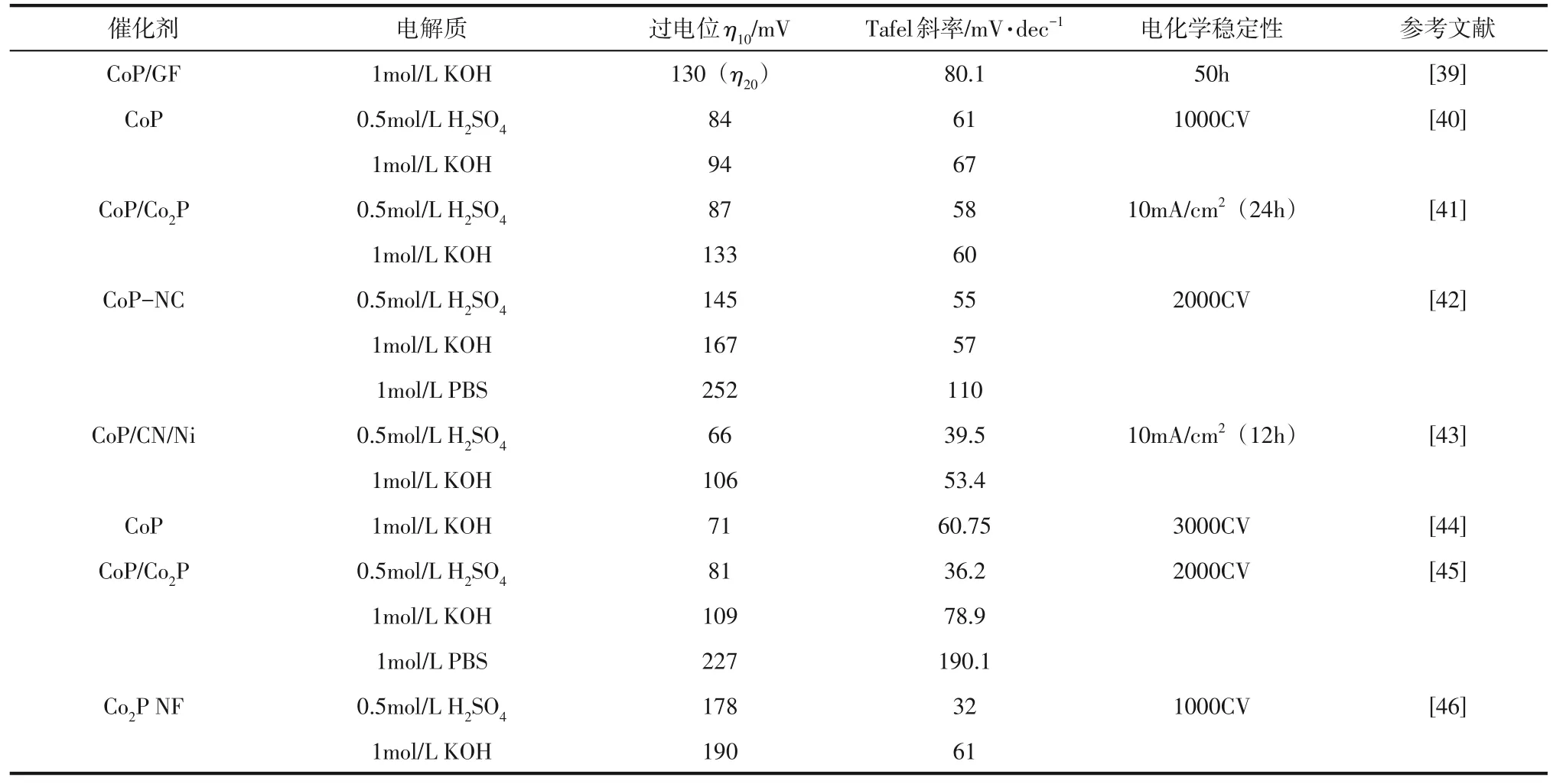
表2 Co基磷化物的HER性能
(3)Fe 作为地球上最低廉、储存量最丰富的过渡金属,其磷化物与唯铁氢化酶的电催化机制相同,是HER的良好催化剂[14,47]。但Fe基磷化物在酸性介质中的稳定性较差,大部分只能应用在碱性环境中,想要实现Fe 基磷化物的广泛应用还需要对其进行较多的研究和改进[48]。表3总结了各种Fe基磷化物的HER性能。
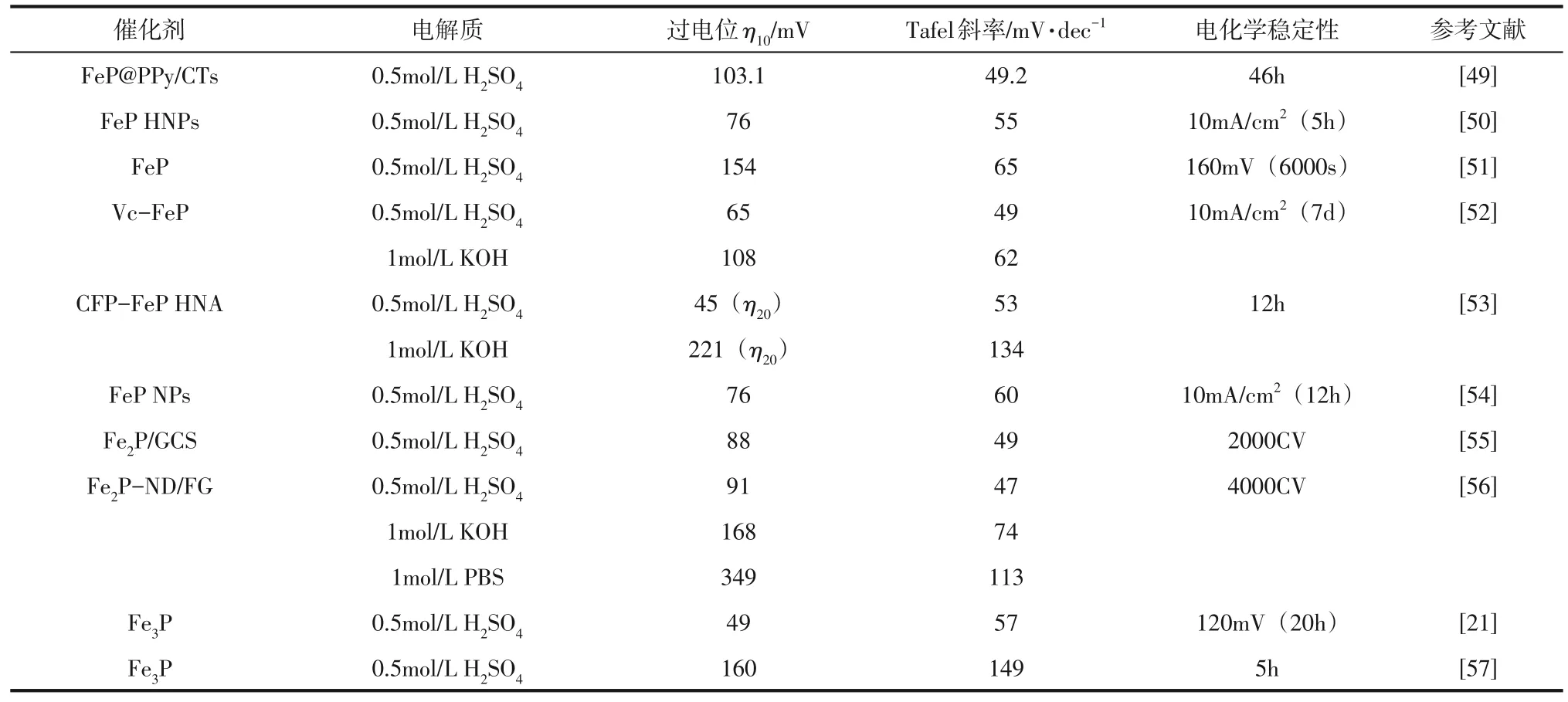
表3 常见Fe基磷化物的HER性能
1.2 导电性能
大多数富磷相的TMPs 是半导体或绝缘体,而富金属相的TMPs 则通常具有优异的导电性能,这是因为富金属磷化物具有丰富的M—M键,含有较多的自由电子,通常会表现出与金属相似的特性,如Mo3P、W3P 和Ni3P 等甚至展现出超导性[13,23]。但P 原子具有较高的电负性,随着P 含量的增加,材料的导电性会逐渐降低。因此,为了平衡TMPs 的催化活性和电导率,需要慎重考虑和控制TMPs 中M 与P 的含量。Pramanik 等[58]合成了具有有序介孔结构的Co2P,在室温条件下电导率为0.4S/cm,是无孔CoP 电导率的37 倍,这归因于Co2P 的富金属相对材料导电性能的提升,以及介孔结构增加了材料的比表面积,为CoP相向Co2P相的转化提供了有力条件。
增强TMPs 的导电性通常可以通过与碳基质结合来实现[59]。在碳基质上负载催化剂材料,不仅能够保证催化剂颗粒的分散性,还能增强电极材料传输电子的能力,从而明显地提高TMPs 的电导率。此外,通过掺杂其他元素来改变TMPs 的电子结构同样可以达到改善材料导电性的目的。
1.3 耐腐蚀性
TMPs 的大多数电化学析氢反应都是在酸性或碱性条件下进行的。中性电解质腐蚀性小,对电解单元的要求低,能够大大降低系统成本,但TMPs在中性条件下HER 过程的动力学较慢,且常用的磷酸盐缓冲溶液(phosphate buffer saline,PBS 溶液)的制备比较复杂,而硫酸钠等新型中性电解质还存在较多的局限性,这就要求应用在HER 中的TMPs 需要具有较好的耐酸碱性[15,60]。因此,改善TMPs 的耐腐蚀性能也是提高其催化活性的一个重要方面。
研究发现,只有CoP和WP2等少数催化剂能够在较宽pH 范围保持高效的HER 催化活性[61-62]。大部分TMPs 具有较好的耐碱性能,然而能在酸性环境中保持稳定的TMPs 材料很少。例如,磷化钛(TiP)、磷化锌(Zn3P2)、磷化镉(Cd3P2)等在水或酸中容易发生水解[63];磷化镍(Ni2P)在酸性溶液中容易被腐蚀[61];磷化铁(FeP)在不同pH溶液中的稳定性较差[63-65]。但是也有一些富含金属离子的磷化物在碱性溶液中容易溶解[61]。
与炭材料结合不仅可以提高材料的导电性,还可以有效提升TMPs 的耐腐蚀性。有研究表明,一些耐腐蚀性较好的金属磷化物通常会形成非晶态薄膜,可以减少晶界处的腐蚀。在合适的制备条件下,碳能够在TMPs 的纳米颗粒外形成碳壳层,起到与非晶态薄膜相似的作用,防止TMPs 被酸碱腐蚀[42,66]。Lin 等[67]将MoP 用 超 薄 的 氮 掺 杂 炭 材 料(nitrogen-doped carbon,NC)进行包覆,制备了MoP/NC球状材料。NC能够对MoP起到有效的保护作用,在酸性和碱性介质中,MoP/NC 材料都比未封装的MoP展现出更好的耐腐蚀性。
1.4 电化学稳定性
在电催化析氢过程中,电极表面的催化剂材料能否在施加电流的情况下保持稳定的催化活性也是十分重要的参考指标。CV 测试和计时电位测量法是评估电化学稳定性的常用方法[15]。
TMPs的电化学稳定性也与其P含量直接相关。Parra-Puerto等[59]成功制备出MxPy/PGC(M=Cr、Co、Ni、Mo、W)催化剂,并在酸性(HClO4)和碱性介质(KOH)中对这些催化剂的电化学稳定性分别进行了测试,结果表明,P 含量最低的Co2P-40[金属磷酸盐负载量为40%(质量分数)]在两种介质中的电化学稳定性均最差。该研究还发现,当电位高于某一特定值时,一些TMPs 的颗粒上会产生氧化物或氢氧化物钝化层,使得催化剂材料能够在碱性环境中保持稳定,这一发现也可以解释大部分TMPs 在碱性介质中稳定而酸性介质中稳定性较差的原因。
需要注意的是,实际电解槽运行需要的高性能非贵金属催化剂要在300mV 的过电位下提供500mA/cm2以上的大电流密度,且要求能维持大电流密度数千小时没有明显衰减[68-69]。而实验室条件下,测试新型析氢催化剂稳定性的工作电位通常较低,电极材料的衰退程度也会有所减弱,无法模拟出在工业应用的高电位下的性能衰退情况[65]。为推进TMPs 的大规模应用,应严格催化剂的稳定性测试条件,尽量贴近实际的工业应用场景。
2 TMPs的制备
TMPs 的合成方法主要有液相反应法、气固反应法、热解还原法和电沉积等。合成方法对TMPs的尺寸、表面结构和颗粒分布起着至关重要的作用。不同的合成条件,如前体的类型、磷化温度和反应时间等,都会对TMPs 的性质与结构造成较大的影响。
2.1 液相反应法
液相反应法是在惰性气体的保护下,将过渡金属、过渡金属盐溶液或金属化合物与有机磷源充分混合生成TMPs 的过程。通过液相反应法得到的TMPs 往往形貌均匀、粒径规则、结晶度好[9,70]。溶剂热法是液相反应的最常见手段[12]。
该方法常用的磷源主要包括三正辛基膦(TOP)、三苯基膦(TPP)和三正辛基氧膦(TOPO)等。TOP 是液相反应最常用的磷源之一,具有作为磷源和稳定表面活性剂的双重作用,对相应磷化物的形态构建有显著的影响[71-72]。目前,TOP已被用于合成空心纳米晶体、纳米线和纳米棒等不同形貌的过渡金属磷化物[59]。Popczun 等[73]在1-十八烯、油胺、三辛基膦中,将Co 纳米颗粒加热至320℃并保持1h,冷却至室温后,用异丙醇清洗,离心后分离得到CoP纳米颗粒。负载在钛基体上的CoP 纳米颗粒展现了良好的HER 催化活性,在酸性条件下达到20mA/cm2的电流密度仅需要85mV的过电位。图4(a)展示了CoP纳米颗粒的制备路径。
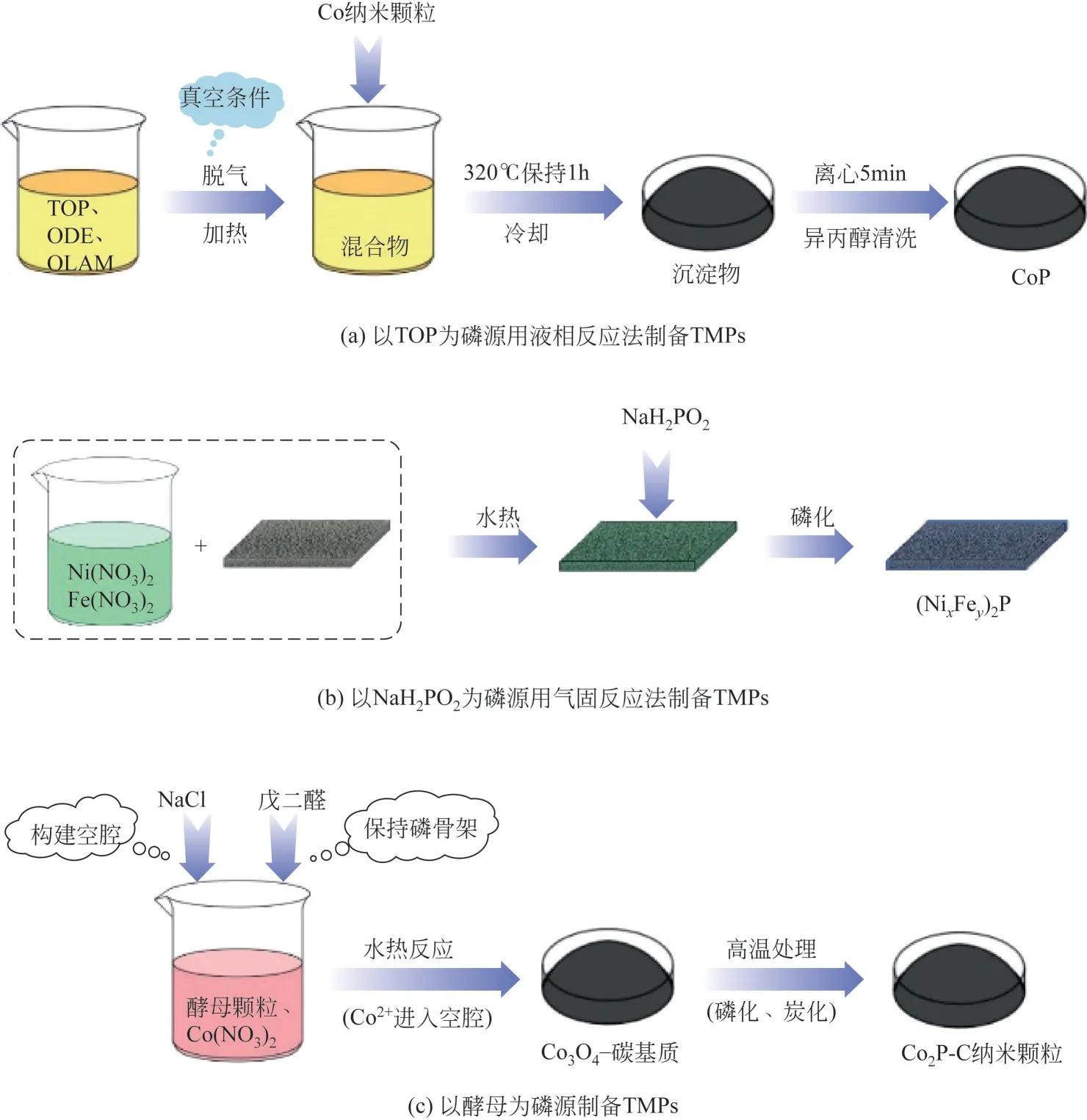
图4 TMPs的制备方法
由于液相反应法是以有机磷作为磷源,并且需要在高温条件下进行,所以反应体系易腐蚀、易燃烧,需要在严格的无氧环境下进行操作[11]。此外,通过液相反应法制备的TMPs 大多为纳米颗粒,需要进一步涂敷在导电基底上才能形成电极材料,这也使得电极的制备步骤变得复杂[71]。
2.2 气固反应法
气固反应法的磷源主要包括次亚磷酸钠(NaH2PO2)和次亚磷酸铵(NH4H2PO2)等无机物。该方法通常在管式炉中进行,磷源在反应过程中产生的磷蒸气或磷化氢(PH3)等,会在保护气的带动下与金属源接触,在特定温度下反应生成TMPs。气固反应制备工艺操作简单,无需高温、高压等苛刻的条件,具有较高的普适性,是最具前景的TMPs制备方法[12,72]。
Li等[7]以NaH2PO2为磷源,用水热法将Ni/Fe 前体生长在泡沫镍上,通过调整Ni2+/Fe3+的比例,得到了一系列不同Ni/Fe比的前体。将磷源和Ni/Fe前体分别置于管式炉的上游和下游,然后在300℃的Ar 气氛下反应2h,制备得到的(Ni0.33Fe0.67)2P 电极在碱性环境下,同时拥有良好的电催化析氢和析氧(oxygen evolution reaction,OER)活 性,达 到50mA/cm2的电流密度,过电位分别需要214mV 和230mV。图4(b)展示了(NixFey)2P电极的制备路径。
此外,红磷等单质磷也是气固反应法的常用磷源。在特定反应条件下,红磷会产生白磷蒸气,从而与金属源反应生成金属磷化物。Jiang等[74]将红磷和Fe(C5H5)2放置于真空密封的石英管中,利用气固反应法制备得到了FeP2/C材料。涂敷在玻碳电极上的FeP2/C 在0.5mol/L H2SO4中展现出了优秀的HER性能。但红磷具有易燃性,因此在使用该种磷源时,需要严格控制反应的真空条件。
一般来说,利用气固反应法制备TMPs,其磷化后样品的粗糙度会略有增加,但其前体的形态可以最大限度地保持不变,该方法有利于构造具有各种特殊形貌结构的TMPs[7,66]。Zhang 等[66]以富含N、S 的Co/Fe 双金属有机骨架为模板,以NaH2PO2为磷源,用低温热解-磷化法制备出CoFeP@NSOC 二元金属磷化物,在400℃下磷化的材料表面变得疏松、粗糙,能够基本保持前体的形貌,但随着磷化温度继续升高,其相变也越来越剧烈,因此利用该方法进行材料制备,需要对磷化的温度进行调控。
气固反应法也存在局限性。次亚磷酸盐在高温下分解通常会产生PH3等剧毒尾气,必须对其进行妥善处理[12,75]。
2.3 热解还原法
热解还原法是在高温的还原性气氛中进行的,该方法多以金属盐为金属源,主要使用的磷源包括磷酸盐和多金属氧酸盐(POMs)等。以磷酸盐为例,在高温中的过渡金属首先被H2等气体还原,而后磷酸根的P—O 键发生断裂并与金属源结合从而形成TMPs。此外,磷酸根还可能会生成挥发性的磷或PH3等气态含磷化合物,这些物质也会参与磷化物的形成[8]。
Chen 等[45]以二己烯三胺五甲叉膦酸(BHMTPMPA)为磷源,采用硝酸钴作为无机金属源,通过热解还原法制备了球形的CoP-Co2P材料。该材料在酸性、碱性和中性介质中均表现出良好的HER 性能,在10mA/cm2的电流密度下,其过电位分别需要81mV、109mV和227mV。
但热解还原法不能很好地控制TMPs 的结构,通过该方法制备得到的TMPs 颗粒往往是不规则且粗糙的[9,76]。为解决这一问题,该制备方法通常与金 属 有 机 框 架 材 料(metal-organic frameworks,MOF)进行结合,以MOF 为模板来限定TMPs 的结构。
2.4 电沉积法
电沉积法主要在室温下进行,其反应条件较为温和,过程中基本不会产生高毒性物质[76]。该制备方法大多以NaH2PO2为磷源,在设定的电位下,溶液中的金属源与磷源会生成TMPs 并附着在工作电极表面。
Roy 等[77]通过电沉积法,将CoP 原位负载在N掺杂的垂直石墨烯纳米山(vertical graphene nanohills,VGNHs)上,制备出了具有多孔纳米花状结构的CoP@N-VGNH 自支撑电极。N 掺杂能够显著提升电子电导率,改善了电极的HER 性能。在酸性和碱性介质中,该电极达到10mA/cm2的电流密度,分别需要29mV 和45mV 的过电位,展现出了优异的HER 性能,尤其是在酸性介质中,CoP@N-VGNH 的过电位低于在相同测量条件下Pt/C(42mV)的过电位。该电极也展现出了良好的稳定性,在60h的工作运行以及1000CV循环后,其结构和功能都没有发生显著变化。
电沉积法相对其他制备方法更为简单、快速,且该方法不需要高温环境,节约了能耗。但电沉积法也存在明显的不足,通过该方法制备得到的TMPs往往结晶性较差,很难具有精细的结构[26]。
2.5 其他方法
除上述常用的制备方法外,TMPs 还可以运用水热法、微乳液法、微波辅助法和室温固相合成法等合成手段[72]。
与易燃、有毒的有机磷和易产生有毒尾气的无机磷相比,环保、无毒的生物质磷源更具发展潜力。例如,Zhang 等[78]以酵母中所含的生物质磷为磷源,以Co(NO3)2·6H2O 为金属源,通过水热反应和高温磷化,制备出了富含中孔的Co2P-C 微球材料。在10mA/cm2的阴极电流密度下,该Co2P-C 复合材料的过电位仅为96mV。这一制备方法能够有效避免复杂前体的使用以及剧毒PH3的产生,且该方法具有普适性,适用于多种磷化物的制备,并按照该方法成功制备出了Mn2P、Ni2P 和Zn3P2材料。图4(c)展示了Co2P-C微球材料的制备路径。
大多数TMPs 的制备都需要高温、严格的惰性或还原性气氛以及有毒尾气的后处理过程。除此之外,由于大多数制备过程中都会涉及到挥发性的含磷物质,容易造成磷的损失,通常还需要过量的磷源来保证磷化的顺利进行,这也造成了磷资源的浪费[8]。针对上述问题,还需要对目前现有的制备方法进行创新性的改进或探索,发展更为简便、安全、有效的TMPs制备方法。
值得提到的是,TMPs 的M/P 化学计量比也对其制备过程至关重要。不同的M/P化学计量比会影响金属磷化物的结晶过程,导致不同计量比的TMPs的结晶温度存在差异[13,79]。表4展示了不同种类的金属磷酸盐热解形成TMPs 的结晶温度及物相组成。
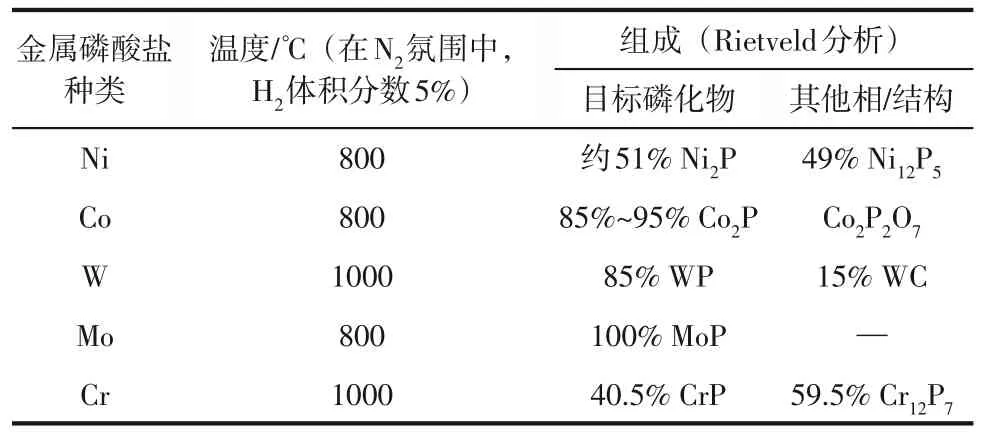
表4 质量分数40%金属磷酸盐在不同温度下热解3h后的物相形成[59]
3 TMPs的改性方法
TMPs 在碱性溶液中具有优异的催化性能,但部分TMPs 在酸性介质中不稳定,这使得TMPs 应用的pH 范围较窄。采用氮掺杂石墨烯、氮掺杂碳纳米管等碳纳米材料对TMPs 进行封装和包覆,能够有效提高TMPs在酸性条件下的稳定性[6,80]。在碱性介质中,通过设计非均相异质结构引入额外的水吸附/解离位点是增强TMPs 催化活性的有效方法。例如,将TMPs与金属氧化物/氢氧化物相结合,后者能够为TMPs提供H2O解离中心[81-83]。但由于金属氧化物的导电性普遍较差,需要注意金属氧化物对材料导电性的影响。由于在不同pH 下的析氢机理有较大差别,因此在酸性和碱性介质中同时实现较高的HER 性能是极具有挑战性的,需要将多种改性方式协调地结合在一起[17]。
此 外,TMPs 非 常 容 易 发 生 表 面 氧 化[30,61,84-85]。Duan 等[85]制备了富含P 空位的v-Ni12P5电极材料,在电极材料的XPS 图谱中,检测到了O—Ni 键和O—P 键的存在,证明Ni12P5发生了表面氧化。同样,Parra-Puerto 等[59]制备出Co2P 催化剂,该材料的STEM-EDX 图谱显示有O 信号的存在,也表明Co2P材料可能存在氧化现象。表面的部分氧化不会影响TMPs 整体的催化性能,但材料表面被过多的氧化物覆盖会导致TMPs 的活性位点减少,材料的导电性也会有所降低。因此,有必要通过元素掺杂或耦合其他材料等手段来防止TMPs的过度氧化[86]。
TMPs 的催化活性会受到M/P 化学计量比、形貌、电子结构、催化环境等诸多因素的影响。针对TMPs 存在的一些问题,可以从以下几个方面对TMPs的性能进行改进。
3.1 元素掺杂
元素掺杂被证明是一种非常有效的改性手段,在TMPs 中掺杂外源元素可以改变其电子结构,对H吸附/解吸自由能进行调控。
3.1.1 金属元素掺杂
在TMPs 中引入其他金属元素能够诱导磷化物中的电荷进行再分配,降低氢吸附自由能,从而提高HER活性。Wang等[87]以ZnCo MOF为模板,通过离子交换、蚀刻、退火和磷化等一系列工艺,制备出了双阳离子掺杂的核壳纳米片结构Fe/Zn-CoP。Fe的引入有助于核壳纳米结构的构建,能够提供丰富的活性位点,Fe的掺杂还加强了电子相互作用,使得CoP的电子结构发生改变,促进了电子的转移速率。Zn元素作为牺牲掺杂剂可以使材料暴露更多的活性位点,提高了HER 活性。该电极在1mol/L KOH 中具有出色的HER 性能,10mA/cm2的电流密度下,HER过电位仅为75mV,且该材料同时还具有良好的OER性能,其过电位仅为267mV。
有研究学者在对OER性能和d带理论进行研究时发现,金属的d带电子结构与氧中间体在催化剂表面的吸附物种结合强度有关,在决定过渡金属合金或化合物性质方面起着重要的作用[88-90]。在HER中,同样观察到了相似的研究规律。Men 等[91]在CoP 中分别掺杂了Fe、Ni、Co、Mn、Cu、Cr、Mo和V 元素,对掺杂后的催化剂在1mol/L KOH 中进行析氢测试,得到的HER 析氢活性顺序依次为V-CoP>Cr(Mo)-CoP>Mn-CoP>Fe-CoP>CoP>Ni-CoP>Cu-CoP,这与掺杂元素的3d 空轨道数的变化顺序一致。DFT计算证明,未经掺杂的CoP的3d 轨道空轨道很少,而掺杂后的材料可以提供更多的空轨道来容纳水的孤对电子,从而促进了吸附过程和解离步骤。
这一掺杂规律也在Man等[92]的研究中得到了证明。在该研究中,Ni 在Ni2P 密排六方晶格中的位置被过渡金属(Fe、Co、Mn、Mo)以单相原子的形式所取代,其金属的结构和形态没有明显变化。在1mol/L KOH中对所制备的催化剂材料进行测试,其HER活性顺序为Ni2P > NiCoP > NiFeP > NiMnP >NiMoP,该顺序与它们在d 轨道上的电子数量排序基本一致,其中Fe和Co的相对位置除外,这可能是因为Co 对H 覆盖率的敏感性强于Fe,从而导致了Fe与Co位置的调换。研究证明,这一排布规律是由于电子排布对H的吸附产生了影响。上述研究结果以及成功的掺杂案例表明,合理运用d带中心理论能够为设计和构建高活性的掺杂电解制氢催化剂提供参考思路[93-94]。
3.1.2 非金属元素掺杂
不同电负性的非金属元素也能够调整TMPs 的电子结构,进而改善材料的HER 活性、导电性和稳定性。Cao 等[95]将硼掺入锚定在碳纳米管上的CoP纳米颗粒中,B的掺杂改变了Co和相邻P原子的局域电子构型和原子排布,优化了活性位点上H吸附和H2解吸的自由能。制备出的B-CoP/CNT 在很宽的pH 范围内都具有优异的HER 活性,在酸性、碱性和中性介质中,分别仅需39mV、56mV和79mV 的过电位就可以达到10mA/cm2的电流密度。B 的掺杂还增强了Co 电子离域能力,提高了电极材料的导电性。
非金属元素的掺杂还可以有效改善TMPs 表面氧化的问题[96]。Kibsgaard 等[97]通过在MoP 中掺杂S制备得到了MoP|S薄膜材料,该材料展现出了比原始MoP更优异的电化学稳定性和催化活性。XPS谱图[图5(a)]表明,暴露于空气中或进行HER 催化后的MoP|S均没有产生PO3-4或SO2-4,证明S的掺杂可以有效防止MoP 的表面氧化,增强了MoP 的稳定性。同时低程度的表面氧化也可能是MoP|S催化活性增强的原因之一,负载量为3mg/cm2的MoP|S 在0.5mol/L H2SO4中 达 到10mA/cm2的 电 流 密 度 仅 需64mV的过电位。图5(b)展示了MoP|S电极的微观结构,MoP|S在钛箔上形成致密的薄膜,薄膜的部分区域发生开裂,产生裂隙。
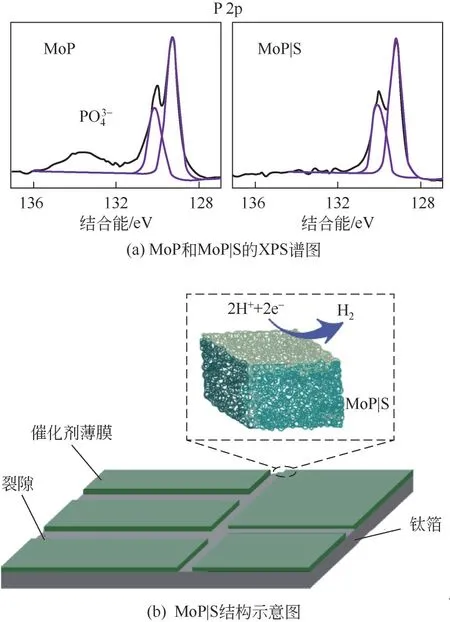
图5 MoP|S的XPS谱图和结构示意图[97]
3.1.3 金属和非金属元素共掺杂
金属和非金属共掺杂也是一种能够有效调节TMPs 催化性能的方法。El-Refaei 等[17]在单质S 的存在条件下对Mn(Fe)、Mo-膦酸盐前体进行还原热解,制备出了Mn(Fe)、S 共掺杂MoP 的MMPS(FMPS)材料。Mn(Fe)的掺杂可以促进MoP 表面的电子富集,改变MoP 的电子结构,使材料产生局部无序的结构,导致其表面产生缺陷和潜在的活性位点,从而促进了质子的吸附并加速了电子转移速率,增强了材料的HER性能和导电性。此外,S 掺杂后的晶体尺寸有所减小,与此结果相对应,测试表明,S的掺杂可以有效提高材料的有效电化学面积(ECSA)。该研究还发现,共掺杂能够有效防止材料氧化,Mn(Fe)的掺杂能够抑制材料表面磷酸盐的形成;S 很难形成含氧化合物,因此S掺杂能够减少材料表面氧化物的产生。这两种材料在不同的pH 下都表现出十分优异的HER 性能,MMPS(FMPS)在酸性介质中达到10mA/cm2的电流密度仅需65mV(68mV)的过电位,而它们在碱性介质中表现出的HER 性能甚至更优,达到相同的电流密度的过电位仅需50mV(51mV)。虽然金属元素的掺杂能够减少磷酸盐的形成,但其更具亲氧性,容易形成氧化物,检测发现,碱性介质中的Mn(Fe)氧化物/氢氧化物数量略多于在酸性介质中的材料,而Mn(Fe)表面氧化物/氢氧化物能够作为水的解离中心,促进了水分子在碱性条件下解离形成吸附态氢的过程。
3.2 构造空位缺陷
缺陷广泛存在于各种晶体中,空位是最常见的缺陷类型,是晶体中的原子或离子离开原位置后留下的空格位。有些空位是晶体生长时内置形成的,有些是通过物理化学等手段诱导形成的。退火是构造空位缺陷的有效方法,高温会导致原子的热运动加剧,其位置发生频繁、随机的变化,从而产生空位缺陷[98-99]。通过调整空位浓度和分布,可以有效地控制晶体的结构、性质和性能。在电催化剂方面,某些特定的空位缺陷位点本身就具有电催化活性;还有一些空位会导致相关原子的配位数减少,激发相邻原子成为新的活性位点。
Zhang 等[84]利用低温自放热的合成方法,通过控制前驱物的质量,制备了结晶度不同、W 空位浓度不同的超细WP纳米颗粒。随着W空位浓度的增加,WP 纳米颗粒逐渐由晶态转变为非晶态。W空位可以激活相邻的P 原子和桥位作为HER 的活性位点;但W 空位或边界会破坏金属骨架的完整性(图6),因此会使WP的导电性变差。由于催化活性与导电性之间存在矛盾,因而需要对WP的空位浓度进行合理调控。低结晶度的LC-WP 处于丰富活性位点和良好导电性之间的平衡点,达到10mA/cm2的电流密度仅需170mV 的过电位。在该项研究中,NaH2PO2作磷源的同时,分解释放的能量还能为WP的合成提供驱动力,使得该合成过程能够在较低的温度下进行,避免了WP颗粒在高温下的过度生长和粗糙化。
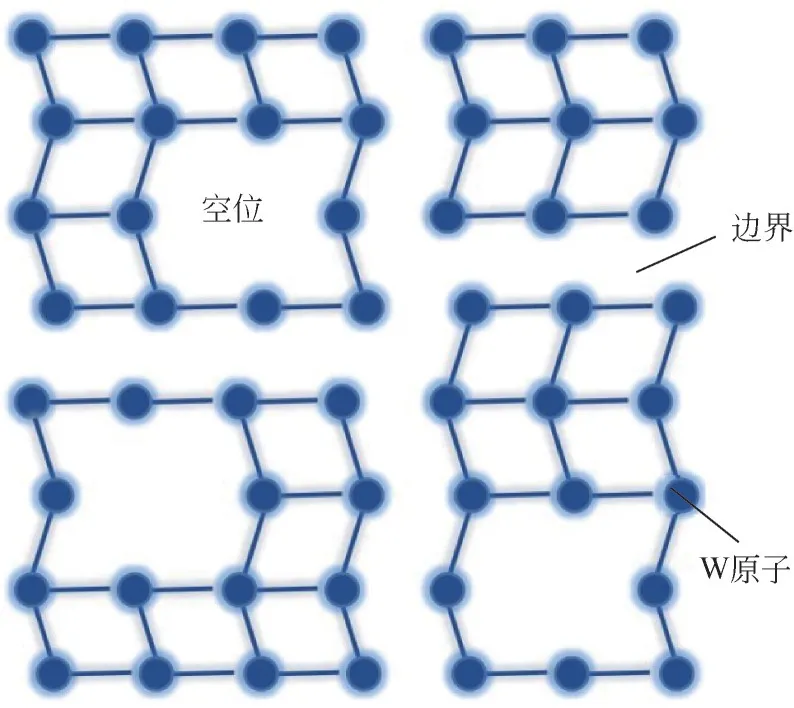
图6 WP含有空位和晶体边界的W—W键模式晶体结构[84]
构造阴离子空位同样能够促进TMPs的HER活性。Duan等[85]通过控制磷源NaH2PO2的用量,采用热退火法在磷化镍(Ni12P5)中构造了磷空位(Pv),制备出的v-Ni12P5电极材料呈纳米线互联的多孔纳米片结构。该研究证明,Pv 可以减弱Ni 的3d 轨道和P 的2p 轨道的杂化,使Pv 附近Ni 和P 原子的电子密度增加,从而促进H*解吸。v-Ni12P5展现出极好的HER 性能,在10mA/cm2的电流密度下,v-Ni12P5的过电位为27.7mV,Tafel 斜率为30.88mV/dec,这一水平优于相同测试条件下的Pt/C基准(32.7mV@10mA/cm2和30.90mV/dec)。
3.3 界面工程
3.3.1 构建非均相异质结构
构建非均相异质结构的目的主要有三点:首先,不同的相能够呈现独特的界面相互作用,这一协同作用可以显著改变界面的物理化学性质;其次,异质结构表面结合处通常会暴露更多的边缘结构,这些结构能够充当H中间体的吸附位点,增加催化剂的活性位点数量;最后,异质结构中不同组分的电负性差异可能会引起不同组分之间发生电子转移,电子再分配会调控材料的电子结构或能带结构,从而提高催化剂的HER活性[83]。
Yang等[100]证明了界面间的相互作用可以促进电催化剂上电子/离子的转移和氢中间体的转运。如图7所示,他们通过电化学沉积技术将铜纳米团簇沉积在泡沫镍(NF)支撑的薄叶状Co2P 上。金属Cu纳米颗粒与半导体Co2P的功函数不同,耦合时形成肖特基结,为使两种材料的费米能级相同,电子会定向从Cu 流向Co2P,这一效应有效地提高了Co2P的导电性能。该材料在1mol/L KOH 中HER 电流密度分别为10mA/cm2和100mA/cm2的条件下,过电位仅为99.7mV 和303.2mV,Tafel 斜率仅48.8mV/dec,表现出了良好的催化活性。该材料还展现出了很好的电化学稳定性,Cu 与Co2P 之间具有强大的附着力,电化学循环中几乎没有发现Cu的损失。

图7 Co2P@Cu/NF结构示意图 [100]
3.3.2 杂化材料
与异质结构不同,杂化材料是在微观尺度发生的均匀混合(图8)。通常情况下,杂化材料的性质并非单纯地介于两种组分之间,而是表现出一种全新的物质特性。有机-无机金属磷化物杂化材料具有丰富可调的组成,是电催化析氢的优良催化剂。Yu等[101]以一维含氮钴基MOF 材料为前体,通过可控的两步热解法将CoP纳米晶体原位限制在氮掺杂的碳纳米线中,制备得到了束状杂化材料(CoP/NCNWs)。该杂化材料具有完整、独特的结构,其催化活性位点丰富,表现出了良好的析氢性能。与在相同条件下制备出的剔除碳物种的磷化钴纳米材料(CoP NPs)相比,在0.5mol/L H2SO4中,CoP/NCNWs 达到10mA/cm2的电流密度所需的析氢过电位仅为95mV,低于CoP NPs材料(125mV)。

图8 非均相异质结构与杂化材料的对比
3.4 耦合炭材料
未经处理的炭材料一般都不具有析氢活性,但碳纳米管、多孔炭和石墨烯等炭材料可以作为析氢电极的良好基底,能够改善电极材料导电性差、难以实现均一纳米尺度的问题。将TMPs 整合到炭材料中,例如用碳纳米管、石墨炭或石墨烯包裹TMPs 纳米颗粒,不仅可以通过导电碳框架提高TMPs 的电导率,促进电子转移,还可以通过碳基底的限制效应来防止纳米颗粒聚集并改善颗粒的分散状态[9,15,20,57,64]。
Zhang 等[102]在多壁碳纳米管(CNT)的侧壁上均匀地包覆了一层尺寸小、结晶良好的MoP 纳米颗粒。MoP纳米颗粒与CNT之间均匀杂化,CNT的加入不仅提高了纳米材料的导电性,而且由于Mo离子与CNT 之间的强相互作用,显著降低了MoP纳米颗粒的团聚和烧结。此外,与CNT 的耦合还有利于MoP 的生长,轻度氧化态CNT 上溶解的Mo配合物与含氧官能团的相互作用有利于Mo 物种在碳纳米管表面的吸附。
在纳米催化剂表面包覆碳基质外壳也是一种防止TMPs 表面氧化的直接、简单和普适的保护方法[47]。Chung 等[103]采用热处理方法将聚多巴胺包覆的氧化铁转化为碳壳层包裹的磷化铁纳米颗粒,如图9所示。扩展X射线吸收精细结构分析、电化学测试与DFT计算一致表明,碳壳层有效减少了FeP纳米颗粒的氧化。此外,碳壳层为活性物质提供了物理和化学保护,能够有效防止磷化过程中纳米颗粒(nanoparticle,NPs)的团聚。

图9 FeP@C的制备流程及结构示意图
与其他碳基材料相比,氮掺杂多孔纳米炭(nitrogen doped porous carbon nanofibers,NPCNFs)具有更强的电负性,在优化HER性能方面具有更大潜力。氮的掺杂会使得相邻碳原子的自旋密度增大,并带有正电荷,最终诱导碳原子成为高效的催化活性位点[79,104]。Wang等[105]采用基于静电纺丝的还原方法,将Ni2P纳米颗粒嵌入氮掺杂的多孔纳米纤维中,制备出了具有一维豌豆状结构的Ni2P@NPCNFs 电极。Ni2P 纳米颗粒在NPCNFs 上分散均匀,不易团聚,纳米颗粒之间具有较低界面电阻,使得电极展现出了优异的导电性能,Ni2P@ NPCNFs的电阻率为5.34Ω·cm,比原始Ni2P的电阻率低104倍左右。与碳基底结合后的Ni2P材料还具有大量的孔隙和较大的比表面积,增强了氢的吸附,实现了高效的电荷转移效率,提高了材料的HER活性。
Zhang 等[79]利用钴离子诱导植酸(PA)和聚苯胺(PANI)前体原位炭化,在柔性炭布上合成了N、P 双掺杂碳纳米结构的柔性自支撑电极(Co2P@NPC/CC)。与传统碳基质一样,N、P 双掺杂碳结构也可以防止Co2P 纳米颗粒在后续热处理过程中发生团聚。双掺杂碳结构还能增强钴离子与碳载体之间的强相互作用,改善了材料的催化活性,该电极在酸性和碱性介质中分别需要116mV和129mV的过电位就能达到10mA/cm2的电流密度。与碳基质的结合也使得该电极具有良好的柔韧性和稳定性,在机械循环应力测试下,其过电位没有发生变化,即Co2P@NPC/CC 电极可以在不同的机械应力下长期保持较高的催化活性。
3.5 微观结构调控
纳米结构设计是在电催化剂中有效添加更多活性位点的有效方法,特别是具有大量纳米孔的核壳纳米结构可以缩短物质扩散距离,加速输运速率,从而提高材料的催化性能。许多HER 催化剂,如六边形MoS2,仅以纳米结构形式存在时才具有活性,而块状的材料则不具有HER 活性,即催化剂的微观纳米结构对催化性能有重要影响[65]。
常见的微观结构包括有空心结构、纳米线、纳米片、纳米管、三明治结构等。通过“模板”(如MOF、光刻掩膜版等)合理设计并构建具有特殊微观结构的TMPs,能够定向调控催化剂或电极的结构和性能[87,106,107]。Zhang 等[108]基于相同的ZIF-67 牺牲模板制备了CoP-HS、CoP2-HS和CoP3-HS三种具有相似中空结构的钴磷化物,其形状类似多孔纳米笼。该结构支持P发挥了重要作用,为HER提供了丰富的活性位点,使得这些钴磷化物表现出优异的HER 性能,达到10mA/cm2的电流密度,分别需要116mV、159mV和170mV的过电位。图10展示了该材料的制备过程,ZIF-67初始为菱形十二面体,由固态转变为中空结构后,其表面出现褶皱。
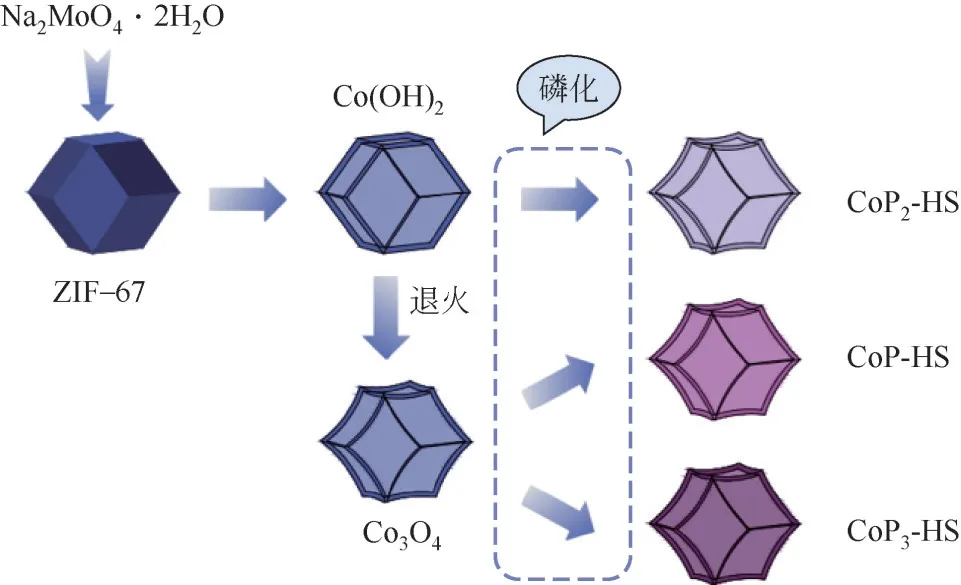
图10 多孔纳米笼的制备示意图 [108]
Wang 等[109]利用NiO-MOF-74 磷化制备了多孔Ni2P纳米片。纳米片的多孔孔道为电子传导提供了途径,促进了电子转移,加速了H2气泡在电极表面的扩散。该材料具有较大的比表面积和出色的导电性能,展现出了良好的HER 性能,多孔Ni2P 纳米片在1mol/L KOH 中,仅需168mV 的过电位就能达到10mA/cm2电流密度。
Liu 等[110]制备的Fe2P@rGO 纳米壁阵列膜具有Fe2P/rGO/Fe2P三明治结构。研究表明,还原氧化石墨烯(reduced graphene oxide,rGO)上的纳米颗粒尺寸比普通氧化石墨烯上的纳米颗粒尺寸小得多。使用合适的载体控制电催化剂颗粒尺寸可有效解决因粒径尺寸过大导致的催化剂层厚、传质能力差、传质过电位高等问题[59]。Lin 的研究表明[110],以rGO 为基底的纳米壁相互缠绕形成开放的空隙,能够加快质量和电子传输速度,这一高度开放的结构使得Fe2P@rGO 的有效电化学面积相较于Fe2P 明显提升,暴露出大量的活性位点。该材料在酸性介质中展现出良好的HER 性能,达到10mA/cm2的电流密度只需101mV的过电位。
3.6 改善材料浸润性
电解水是典型的析气反应,如果气体产物不能尽快脱离电极界面,就会使得活性位点被气泡堵塞,不利于电化学反应的进行。提高材料的疏气性,促进电极表面气泡的释放,能够提高电极在高电流密度下的析氢能力。通过调节材料的浸润性及电极的宏观形貌,可以对水下气泡的行动路径实现精确操控[111]。如图11 所示,良好的析氢电极应该具有优异的亲水性和疏气性,亲水性能够使得溶液在电极表面迅速铺平,增强两相之间的接触,促进电荷转移;而疏气性可以加速气泡从电极表面脱离。一般来说,具有亲水性的电极往往也具备疏气性,但该规律并不适用于所有的材料。
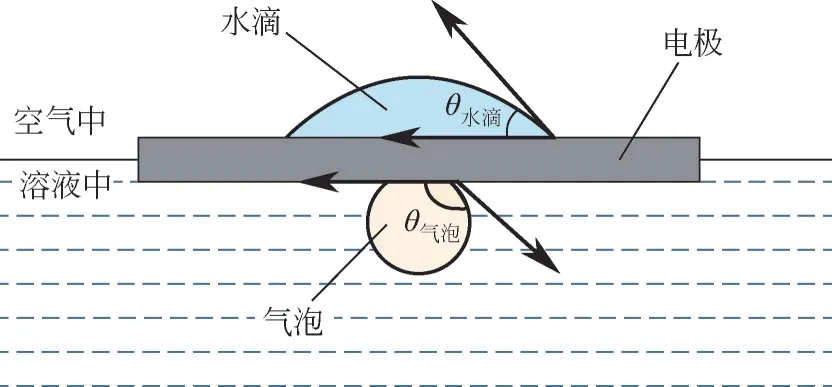
图11 同时具有亲水性和疏水性的电极材料
浸润性由材料的表面结构特性和化学成分共同决定。构造纳米阵列是改善材料浸润性的常见方式,制备具有规律排布结构的纳米阵列自支撑电极更容易实现表面亲/疏水工程[68]。
具有超亲水性能的材料往往也具有超疏氧性能,超疏氧性会导致“气泡破裂效应”的产生,能够加速气泡释放[112]。Chen 等[113]制备出了具有超亲水和超疏氧性质的NF@CoxP 材料。未经处理的泡沫镍(nickel foam,NF)的水滴接触角为66.59°,而NF@CoxP 的接触角为0°,这证明NF@CoxP 具有超亲水特性,能够使得水在电极表面迅速吸附和铺平,提高了CoxP 和电解质之间的离子转移速率。超厌氧性使得生成的H2气泡在电极表面的快速释放,防止了活性位点被气泡覆盖。
运用“模板法”来构造纳米阵列也是非常有效的。Kim 等[106]以MOF 为模板,直接在NF 上构建了具有3D“头”“身”结构的CoP@Ni-CoP “蘑菇”状电极。在磷化过程中引入的亲水性磷酸基团使得该电极具有高亲水性,其水滴接触角为0°,能够促进气泡的快速释放。在改性过程中,MOF 只生长在纳米阵列的尖端,而Ni 只在MOF 处发生了选择性修饰,这一特殊结构使得该材料的有效电化学面积明显增大。CoP@Ni-CoP 电极同时拥有优异的HER 和OER 性能,在碱性介质中达到10mA/cm2的电流密度分别只需63.4mV 和281.0mV 过电位。图12展示了CoP@Ni-CoP的制备流程。
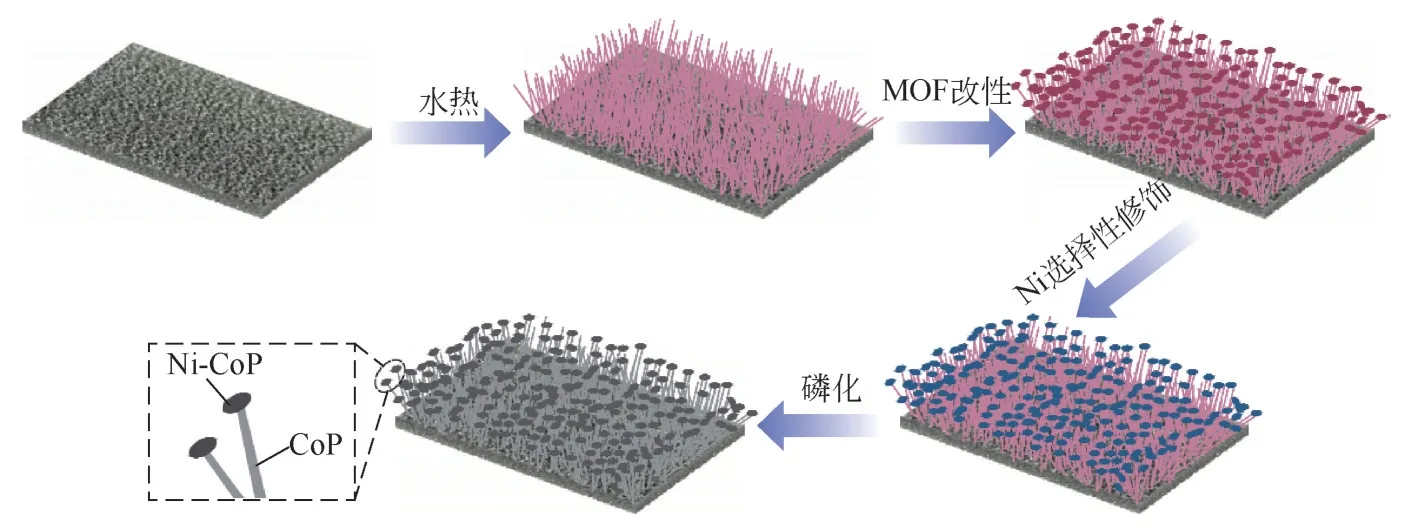
图12 CoP@Ni-CoP制备流程[106]
3.7 其他改性方法
(1)催化剂材料表面重构 电化学过程可能会导致电催化剂发生还原、非晶化、氢吸附能力改变和元素浸出等变化[62,114]。决定材料稳态性能的往往是重构表面而非原始表面,因此电解过程中电催化剂的表面重构是非常重要的[47,115]。Su等[116]对CoP纳米片进行原位电化学活化,研究了表面重构对催化剂性能的影响。CoP 经过活化后,表面的P原子被浸出,导致相邻的Co 原子暴露。这种暴露的Co非常活跃,非常容易与电解质中的OH-结合形成Co(OH)x,最终形成了稳定的Co(OH)x@CoP 杂化结构。表面重构导致CoP纳米片的形貌和组成发生了不可逆变化,Co(OH)x@CoP杂化界面的协同作用使得催化剂表面的羟基基团有所增加,促进了水的解离,改善了材料的HER 活性。在碱性介质中,要达到10mA/cm2的电流密度,原始CoP 纳米片需要的过电位为180mV,而经过表面重构后的Co(OH)x@CoP所需要的过电位仅为100mV。
(2)利用非晶态TMPs 修饰催化剂材料 由于催化剂表面原子的排列差异而引起的面效应也对HER的研究具有重要意义。非晶态物质的原子排列是近程有序和长程无序的,在结构上偏离了晶体材料,因此它具有广泛可调的组成、均匀的组成和较多的不饱和配位点,被广泛应用于催化反应。目前的研究大多集中在晶态TMPs上[13],但从暴露活性位点、调节表面积等方面来看,非晶态TMPs 展现出来的性能更为优越[86,102]。Zhang等[117]将非晶态的Co2P修饰在Co-多金属氧酸盐(Co-POM)和导电磷化钴电极上,在低温条件下制备得到了Co2P@ Co2P/Co-POM/NF电极。非晶态Co2P为电子传输提供了通道,加快了导电磷化钴的电子传输速度,降低了电荷转移电阻。这一设计使得材料具有良好的导电性,增加了边缘活性位点的数量,提高了电极的HER 性能。在碱性介质中,Co2P@ Co2P/Co-POM/NF电极达到50mA/cm2的电流密度仅需130mV的过电位。
(3)拓扑转化法 这是改善HER 催化剂活性的一个新方法。拓扑转化可以通过使用封端剂或还原剂来改变晶体结构、相和表面形貌,使得晶体颗粒尺寸实现最小化。经拓扑转换处理后的纳米粒子的排列更加有序,能够提高催化剂的活性和稳定性[86]。除上述改性方法外,利用光、磁和电场等外部辅助手段也能够改善TMPs的HER性能[118]。
4 结语与展望
本文对TMPs 的性质、制备方法以及改性方式进行了综述。TMPs 由于具有优越的催化性能、低廉的成本和较好的稳定性,已经得到了广泛的研究。TMPs中的M/P化学计量比对其性质尤为重要,想要得到性能良好的TMPs,需要在制备过程中合理调控M/P 比,必要时还应辅以合适的改性手段。综合各类文献的研究结果可以看出,不同调控TMPs 性能的手段具有部分矛盾或冲突的地方,因此对于TMPs 材料的改进方法需要综合考虑,在催化活性、导电性、耐腐蚀性和电化学稳定性等性质中间寻找一个平衡点,从而得到能够满足催化性能要求的最优材料。总体而言,TMPs 析氢材料在电解水制氢领域的研究与应用越来越受到关注,当前及未来的研究重点主要体现在以下几个方面。
(1)新型磷源的开发 目前制备磷化物常用的传统磷源具有明显局限性,如危险性较高、易对环境造成较大危害等。发展新型磷源对促进磷化物在析氢领域的大规模应用具有重要作用。
近年来,离子液体的兴起为新型磷源的开发拓宽了道路。以磷基离子液体为介导,通过改变金属源阴离子的种类,能够成功实现对金属磷化物的形态、尺寸和晶相的调控[119]。在开发新型磷源的同时,相应的制备方法也需要同步更新或改进。采用微波加热工艺对磷基离子液体与金属源进行处理,能够在极短的时间内实现金属磷化物的成功制备。与传统制备方法相比,以离子液体为磷源的微波加热法具有反应快速、能耗较低、无剧毒尾气等优点。
此外,生物磷也是极具发展前景的一类磷源。以微生物细胞或某些生物质为磷骨架,利用特定方法将金属元素锚定在磷骨架中,能够以更为绿色、环保的形式实现金属磷化物的成功制备。
(2)测试标准化 大规模工业化应用的电极,需要实现在大电流密度、长运行时间、极端碱性环境和较高温度等条件下的稳定、持续产氢。但目前对于TMPs 的电催化性能测试还停留在实验阶段,且其相关测试指标没有统一的评价标准。这些问题不但阻碍了TMPs 的规模化应用,还在一定程度上影响了相关科研工作者对于材料性能的评判与比较。因此,实现电化学测试指标的标准化,对于TMPs材料的应用与普及具有重要的现实意义。
(3)晶面调控 相关研究表明,TMPs 的不同晶面所展现的电催化性能也不尽相同,例如,Ni2P的(001)晶面相较于其他晶面更有利于析氢反应的发生[96,120]。因此,尽可能地暴露出高析氢晶面,或制备出具有相应晶面的单晶材料,为改善TMPs 材料的电催化析氢性能提供了新的思路。利用这一规律,通过对材料晶面的判断,还能够实现对于TMPs催化活性的预测。然而,目前对于TMPs的晶面调控研究还较为欠缺,需要进一步的探索与实验。
(4)电极基底的选择 电极的基底在调节TMPs 材料的催化性能方面起着极其重要的作用。基底的本征性质,如导电性、稳定性等,对电极整体性能存在明显影响。此外,基底材料与催化剂产生的协同效应,会改变电子间的相互作用或者导致具有催化活性界面的形成。常见的基底主要包括炭材料、金属以及其他导电材料,应用较多的有炭布(CC)、金属泡沫(如NF)、金属网(如钛网)、金属箔(如钼箔)、氟掺杂氧化锡(FTO)等。近年来,黑磷也逐渐被应用为电催化基底,该物质具有波层状结构,与石墨类似,是白磷和红磷的同素异形体,研究发现,以黑磷为基底也可以有效提升TMPs的析氢性能[20,121-122]。
猜你喜欢
电镀与精饰(2022年10期)2022-10-14
食品安全导刊(2021年20期)2021-11-28
电镀与环保(2017年6期)2018-01-30
电镀与环保(2017年5期)2017-12-19
电镀与环保(2017年3期)2017-06-23
电镀与环保(2016年2期)2017-01-20
现代工业经济和信息化(2016年12期)2016-05-17
中国资源综合利用(2016年4期)2016-01-22
广州大学学报(自然科学版)(2015年4期)2015-12-23
电源技术(2015年2期)2015-08-22
