
狂犬病病毒基质蛋白74位氨基酸位点对其线粒体定位的影响
2023-07-31 09:05:52王姝捷周宏婧许运斌
中国人兽共患病学报 2023年12期
张 曦,王姝捷,周 俊,高 琛,刘 琴,周宏婧,许运斌
据WHO报道,狂犬病在全世界每年可造成近6万人死亡,而绝大多数疫情发生在亚洲和非洲等发展中国家[1]。在历史上的一段时间,我国是狂犬病高发地区,但随着政府监管系统的改进及动物咬伤人群能够得到及时的预防性医疗救治,我国报告的人狂犬病病例从2007年的3 300例稳步下降到2020年的202例[2]。尽管如此,国家卫健委每年公布的全国法定传染病疫情概况显示,狂犬病一直以来是我国法定乙类传染病报告死亡数居前5位的病种,依然对我国公共卫生安全构成严重威胁。而在我国,约95%的人狂犬病是由患病犬咬伤所致[1]。因此,继续通过加强犬群管理、大规模免疫以及流行病学监测来控制犬狂犬病,是预防和控制人狂犬病的关键。然而,狂犬病一旦发病,死亡率几乎为100%,仍无治愈希望。
狂犬病是由狂犬病病毒(rabies virus,RABV)引起的几乎所有温血动物都易感的人兽共患病。RABV是不分节段的单股负链RNA病毒,其基因组长度大约12kb,编码核蛋白(N)、磷蛋白(P)、基质蛋白(M)、糖蛋白(G)和聚合酶蛋白(L)等5种病毒结构蛋白。其中M蛋白是一种由202个氨基酸组成的多功能蛋白,在调控病毒转录与复制、促进病毒粒子组装和出芽、诱导细胞凋亡和影响病毒致病力方面发挥着重要作用[3-11]。在调控细胞凋亡方面,昝洁等研究发现,RABV毒株CVS-11感染可激活依赖和非依赖caspase的线粒体凋亡途径,从而诱导小鼠神经瘤母瘤细胞N2a、幼仓鼠肾细胞BHK-21、人胚肾细胞293T凋亡[8]。该研究还发现,病毒M蛋白可部分定位至线粒体并诱导线粒体途径的凋亡;而且,该蛋白中通过软件预测的疏水性α螺旋区域(67-79 aa)具有严格的线粒体靶向作用,单独转染表达具有诱导细胞凋亡的能力,而74位aa的突变(H→P)则可破坏α螺旋并使得该区域失去定位于线粒体和诱导凋亡的能力[8]。尽管如此,74位aa的突变(H→P)是否可完全破坏全长病毒M蛋白定位至线粒体的能力,或者疏水性α螺旋区域(67-79aa)对于病毒M蛋白定位至线粒体是否起着决定作用尚不清楚。
本研究首先对哺乳动物细胞慢病毒表达载体pLVX-Puro-MCS(多克隆位点)进行改造,分别构建骨架载体pLVX-puro-MCS-V5-APEX2和pLVX-Puro-APEX2-V5-MCS,接着设计引物分别将RABV野生型病毒M基因及其74位aa的突变基因(H→P)克隆至上述骨架载体MCS区域。对所构建载体经测序验证成功及免疫印迹鉴定表达成功后,将载体转染293T细胞后进行亚细胞定位分析,以确定74位组氨酸对全长病毒M蛋白定位至线粒体能力的影响,有助于解析M蛋白的生物学功能,为狂犬病复制和致病机制研究提供理论依据。
1 材料与方法
1.1 材 料
1.1.1 细胞培养 人胚肾上皮细胞系293T、人神经母细胞瘤细胞SK-N-SH培养于含5%胎牛血清的DMEM/F12培养基中。采用SK-N-SH细胞培养狂犬病病毒固定毒株CVS-11(二级生物安全操作水平毒株),测定病毒滴度并分装,-80 ℃保存于实验室。
1.2 方 法
1.2.1 真核表达载体构建 委托广州辉骏/辉腾生物科技股份有限公司构建真核表达载体。
1.2.1.1 pLVX-puro-MCS-V5-APEX2相关载体的构建 1)首先全基因合成M(67-79 aa)-V5-APEX2基因相关序列,结合pLVX-Puro-MCS载体多克隆位点酶切位点序列,设计引物如下:上游引物:5′-GGTACCGCGGGCCCGGGATCCATGGGCAAG-CCCATCCCCAAC-3′,下划线为BamHⅠ酶切位点;下游引物:5′-TACCCGGTAGAATTATCTA-GATTAGGCATCAGCAAACCCAA-3′,下划线为XbaI酶切位点。采用上述引物以全基因合成序列为模板对V5-APEX2基因序列进行PCR扩增。95 ℃预变性2 min;95 ℃变性20 s,60 ℃退火30 s,72 ℃延伸1 min,30个循环;72 ℃彻底延伸3 min。采用BamH I和XbaI限制性内切酶分别对PCR回收产物及pLVX-Puro-MCS进行双酶切,DNA凝胶回收相应条带后进行连接,转化Stbl3感受态细胞,挑取单克隆进行PCR鉴定,选取阳性单克隆进行测序验证,从而成功构建pLVX-puro-MCS-V5-APEX2载体。2)在pLVX-puro-MCS-V5-APEX2骨架载体的基础上,结合其多克隆位点酶切位点序列、野生型M基因序列及M(H74P)突变基因序列,设计特异性引物进行PCR、双酶切、多片段拼接克隆、转化、测序等,从而成功构建pLVX-puro-M-V5-APEX2及pLVX-puro-M(H74P)-V5-APEX2载体。其中pLVX-puro-M-V5-APEX2载体的构建采用2对特异性引物;pLVX-puro-M(H74P)-V5-APEX2载体的构建采用3对特异性引物(具体引物信息见表1)。

表1 pLVX-puro-M-V5-APEX2和pLVX-puro-M(H74P)-V5-APEX2载体构建相关引物信息
1.2.1.2 pLVX-Puro-APEX2-V5-MCS的构建 1)结合pLVX-Puro-MCS载体多克隆位点酶切位点序列,设计引物如下:上游引物:5′-CTACCGGACTCAGATCTCGAGATGGGAAAGTCTTACCCAA-CTGTG-3′,下划线为XhoI酶切位点,下游引物:5′-GTACCGTCGACTGCAGAATTCGGTGCTG-TCCAGGCCCAGCA GGGGGTTGGGGA-TGGG-CTTGCCGGCATCAGCAAACCCAAGCT(含有V5标签基因序列)-3′,下划线为EcoR I酶切位点。采用上述引物以全基因合成序列为模板对APEX2基因序列进行PCR扩增。95 ℃预变性2 min;95 ℃变性20 s,60 ℃退火30 s,72 ℃延伸1 min,30个循环;72 ℃彻底延伸3 min。采用XhoI和EcoR I限制性内切酶分别对PCR回收产物及pLVX-Puro-MCS进行双酶切,DNA凝胶回收相应条带后进行连接,转化Stbl3感受态细胞,挑取单克隆进行PCR鉴定,选取阳性单克隆进行测序验证,从而成功构建pLVX-Puro-APEX2-V5-MCS载体。2)在pLVX-Puro-APEX2-V5-MCS骨架载体的基础上,结合其多克隆位点酶切位点序列、野生型M基因序列及M(H74P)突变基因序列,设计特异性引物,以上述构建成功的野生型M基因或M(H74)突变基因载体为模板,进行PCR、双酶切、连接、转化、测序等,从而成功构建pLVX-puro-APEX2-V5-M及pLVX-puro-APEX2-V5-M(H74P)载体。其中pLVX-puro-APEX2-V5-M载体的构建采用1对特异性引物:上游引物:5′-CTACCGGACTCAGATCTC-GAGATGGGAAAGTCTTACCCAA-CTGTG-3′,下游引物:5′-GAATTCGGTGCTGTCCAGGCCCAGCAGGGGGTTGGGGATGGGCTTGCCGG-CATCAGCAAACCCAAGCT-3′。pLVX-puro-APEX2-V5-M(H74P)载体的构建采用1对特异性引物:上游引物:5′-GGCCTGGACAGCACC-GAATTCATGAACGTTCTACGCAAGAT-A-3′,下游引物:5′-TTATCTAGAGTCGCGGGATCCTCATTCTAG-AAGCAGAGAAGAGTC-3′。
1.2.2 免疫印迹实验 将1.2.1中构建成功的表达载体转染SK-N-SH,转染48 h后采用RIPA(中)裂解液(购自上海碧云天生物技术有限公司)裂解细胞,收获细胞总蛋白,按比例加入SDS 5×蛋白上样缓冲液(购自上海碧云天生物技术有限公司),100 ℃煮沸10 min。接着对煮沸过的蛋白样品进行4 ℃、12 000 r/m离心10 min,取蛋白样品上清进行SDS-PAGE蛋白电泳,程序为80 V恒压120 min,至目的条带分离充分;300 mA、150 min冰浴条件下进行湿转。湿转成功的NC膜用5%的牛血清白蛋白(BSA)4 ℃过夜封闭,弃去5%BSA后用1×PBST洗膜3次;加入一抗稀释液稀释好的一抗(抗V5标签抗体或M蛋白鼠多抗),4 ℃孵育过夜;1×PBST洗膜3次,加入稀释的HRP标记的羊抗兔(1∶1 000)或羊抗鼠IgG(1∶1 000),室温孵育4 h;1×PBST洗膜3次,每次15 min。最后采用化学发光仪上进行ECL显色拍照。如此,鉴定所构建的真核表达载体的表达情况。
1.2.3 荧光染色实验 将1.2.1中构建的真核表达载体或突变体分别单独转染细胞或分别与pDsRed2-mito质粒共转染细胞后,经Mito-tracker染料染色、细胞固定和免疫荧光染色,基本步骤:细胞转染48 h后弃掉培养基,加入用培养基稀释的MitoTrackerTMDeep Red FM染料100 μL,在37 ℃细胞培养箱孵育35 min,接着对染色后的细胞进行4%多聚甲醛、0.2%Triton X-100固定和通透,加入5%脱脂奶稀释的V5标签抗体37 ℃孵育1 h,PBS清洗后加入5%脱脂奶稀释的FITC标记的羊抗兔IgG进行孵育,37 ℃孵育1 h;或经细胞固定和免疫荧光染色,基本步骤:对转染48 h后的细胞进行4%多聚甲醛、0.2%Triton X-100固定和通透,加入5%脱脂奶稀释的V5标签抗体37 ℃孵育1 h,PBS清洗后加入5%脱脂奶稀释的FITC标记的羊抗兔IgG进行孵育,37 ℃孵育1 h。最后用PBS进行清洗,采用激光共聚焦显微镜拍照观察APEX2标记的M蛋白或M(H74P)突变蛋白与线粒体的共定位情况。
2 结 果
2.1 真核表达载体构建 经广州辉骏/辉腾生物科技股份有限公司对所构建载体进行测序,结果显示载体中V5标签序列、APEX2基因序列、野生型M基因及M(H74P)突变基因序列正确,且未发生移码,如此成功构建出真核表达载体pLVX-puro-MCS-V5-APEX2、pLVX-puro-M-V5-APEX2、pLVX-puro-M(H74P)-V5-APEX2、pLVX-Puro-APEX2-V5-MCS、pLVX-puro-APEX2-V5-M及pLVX-puro-APEX2-V5-M(H74P)。
2.2 M蛋白C端与APEX2融合表达鉴定 将质粒pLVX-puro-MCS-V5-APEX2、pLVX-puro-M-V5-APEX2、pLVX-puro-M(H74P)-V5-APEX2(分别缩写为V5-APEX2、M-V5-APEX2、M(H74P)-V5-APEX2)分别转染至SK-N-SH细胞,转染48 h后收获总蛋白样品,采用V5标签抗体进行免疫印迹可检测各融合蛋白(图1A),采用M蛋白鼠多抗进行免疫印迹可检测M-V5-APEX2、M(H74P)-V5-APEX2融合蛋白(图1B),条带大小与预期相符。

图1 免疫印迹检测M蛋白C端与APEX2的融合表达情况
2.3 M蛋白C端与APEX2融合表达后与线粒体共定位关系 将构建成功的APEX2与M蛋白C端融合表达的慢病毒真核表达载体(V5-APEX2、M-V5-APEX2、M(H74P)-V5-APEX2),分别单独转染细胞或分别与pDsRed2-mito质粒共转染细胞后,经Mito-tracker染料染色、细胞固定和免疫荧光染色,或经细胞固定和免疫荧光染色,最后进行激光共聚焦显微镜拍照观察,初步发现APEX2与M蛋白C端融合表达之后并未影响M蛋白的线粒体定位能力(图2A、2B),同时还初步发现74位aa的突变(H→P,H74P)也并未使得全长病毒M蛋白完全失去定位至线粒体的能力(图2A、2B)。
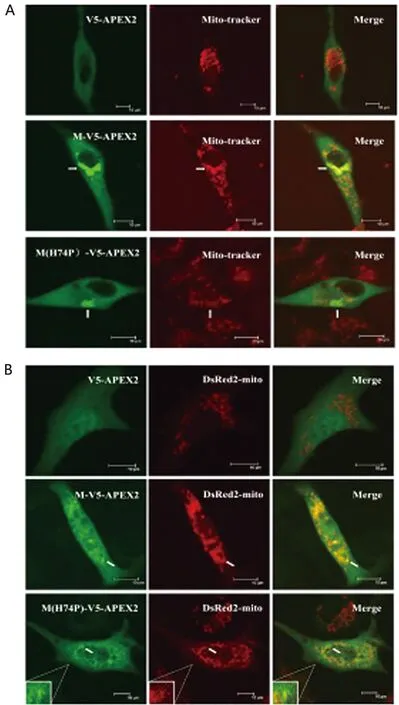
A:Mito-tracker染料染色后进行免疫荧光染色,并结合激光共聚焦显微镜拍照观察检测APEX2标记的M蛋白与线粒体的共定位关系;B:免疫荧光染色结合激光共聚焦显微镜拍照观察检测APEX2标记的M蛋白与线粒体的共定位关系。
2.4 M蛋白N端与APEX2融合表达鉴定 将质粒pLVX-Puro-APEX2-V5-MCS、pLVX-puro-APEX2-V5-M及pLVX-puro-APEX2-V5-M(H74P)(分别缩写为APEX2-V5、APEX2-V5-M、APEX2-V5-M(H74P))分别转染至SK-N-SH细胞,转染48 h后收获总蛋白样品,采用V5标签抗体进行免疫印迹可检测各融合蛋白(图3A),采用M蛋白鼠多抗进行免疫印迹可检测APEX2-V5-M、APEX2-V5-M(H74P)融合蛋白(图3B),条带大小与预期相符。

图3 免疫印迹检测M蛋白N端与APEX2的融合表达情况
2.5 M蛋白N端与APEX2融合表达后与线粒体共定位关系的检测 本研究还构建了APEX2与M蛋白N端融合表达的慢病毒真核表达载体分别为APEX2-V5、APEX2-V5-M、APEX2-V5-M(H74P),分别单独转染细胞或分别与pDsRed2-mito质粒共转染细胞后,也同样发现APEX2与M蛋白N端融合表达之后也未影响M蛋白的线粒体定位能力(图4A、4B),同时也发现74位aa的突变(H→P,H74P)也并未使得全长病毒M蛋白完全失去定位至线粒体的能力(图4A、4B)。
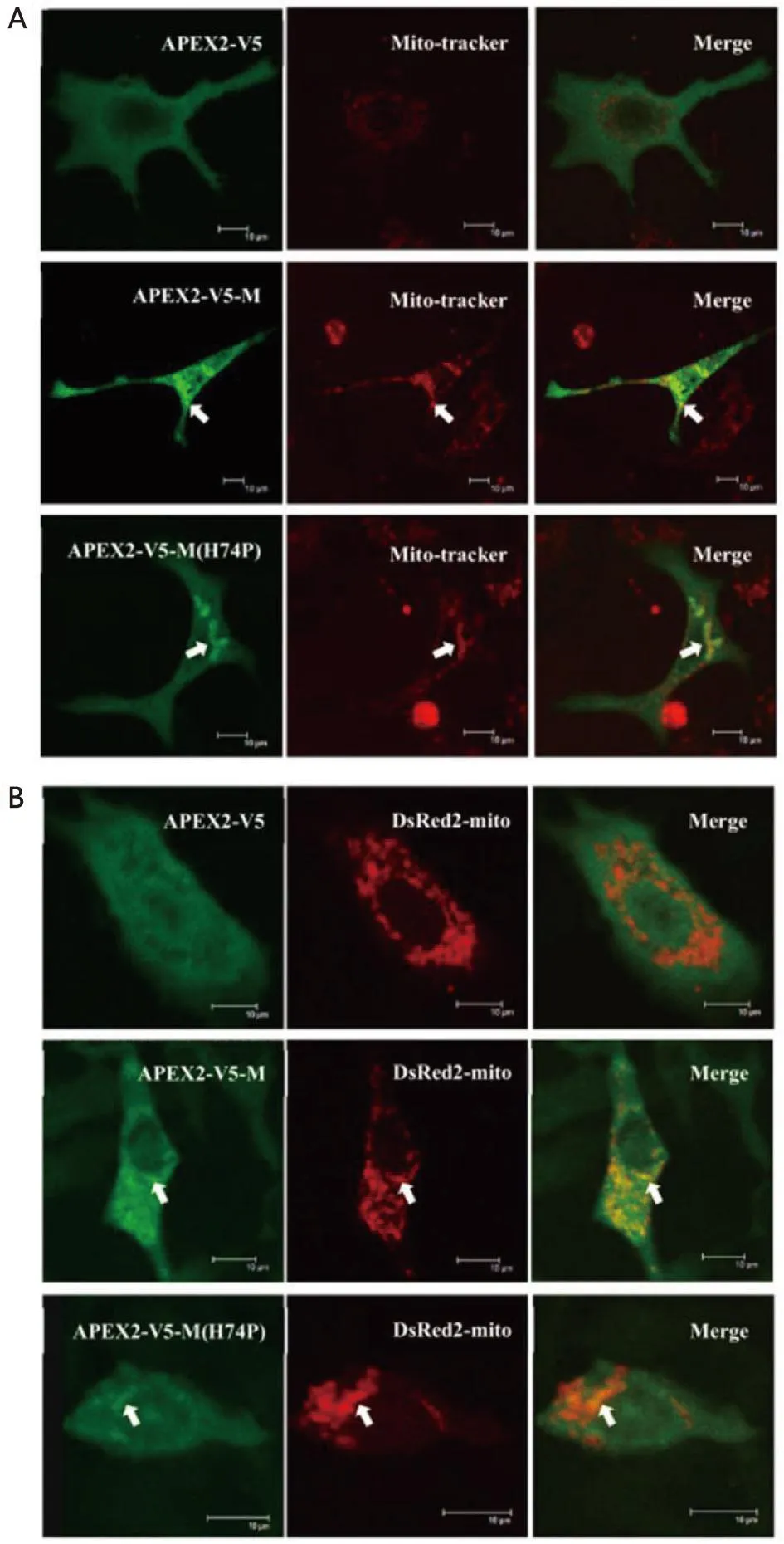
A:Mito-tracker染料染色后进行免疫荧光染色,并结合激光共聚焦显微镜拍照观察检测APEX2标记的M蛋白与线粒体的共定位关系;B:免疫荧光染色结合激光共聚焦显微镜拍照观察检测APEX2标记的M蛋白与线粒体的共定位关系。
3 讨 论
据报道,在感染乳鼠、免疫抑制成年小鼠及具有免疫功能的成年小鼠后,RABV毒株CVS-11可诱导脑细胞显著的凋亡效应[12-13]。进一步研究发现,CVS-11毒株的复制可诱导小鼠神经母细胞瘤细胞Bax相关、caspase依赖的凋亡[14]。一直以来,RABV病毒G蛋白被公认为在诱导细胞凋亡中起着重要作用[15-16]。然而,有研究报道称,含有非致病性RABV毒株G基因的病毒才能诱导细胞凋亡,而含有固定毒株CVS-11 G基因的病毒则没有此效应[17]。那固定毒株CVS-11如何诱导细胞凋亡?在本研究之前,昝洁等研究发现RABV毒株CVS-11病毒M蛋白可部分定位至线粒体并诱导细胞凋亡,其中67-79 aa这段疏水性的α螺旋区域具有严格的线粒体定位作用,并可诱导细胞凋亡[8],但截至目前具体分子细节仍不清楚。在本研究中,我们证明了M蛋白的N端或C端与APEX2融合表达均未最终改变M蛋白定位至线粒体的能力。由此说明,74位aa的突变(H→P)虽能破坏疏水性α螺旋区域(67-79 aa)定位至线粒体的能力[8],但是未能完全破坏全长病毒M蛋白定位至线粒体的能力,或者疏水性α螺旋区域(67-79aa)对于病毒M蛋白定位至线粒体未能起着决定作用,暗示着病毒M蛋白极有可能还存在其他序列可专一定位至线粒体。这有待后续继续深入研究。
此外,在本研究中,我们构建的真核表达载体均携带有APEX2标签。APEX2是一种APEX改进型的抗坏血酸过氧化物酶,能催化生物素-苯酚和过氧化氢(H2O2)之间的反应,形成一种能迅速与附近的氨基酸共价结合的生物素偶联的苯氧自由基[18-19]。APEX2邻近标记(APEX2-based proximity labeling)半径小于20 nm,这种标记反应在活细胞内只需1 min即可在原位通过这些自由基与邻近蛋白质上电子密度较高的氨基酸(如Tyr、Trp、His和Cys等氨基酸)结合而使得邻近蛋白质被高效地生物素化,接着采用链霉亲和素磁珠对生物素化的蛋白质进行富集和质谱鉴定,该方法相比其他的邻近标记法耗时更短,更有利于研究特定实验条件下某个时间点的瞬时互作蛋白质组学信息。随着APEX2标记技术的成熟和发展,运用APEX2邻近标记结合SILAC、质谱检测的方法筛选并鉴定与定位于细胞器的病毒蛋白互作的未知蛋白,同时运用APEX2标记结合透射电子显微镜成像的方法解析这些病毒蛋白在细胞器中所处具体位置和拓补结构,这将对全面深入阐明病毒蛋白定位于细胞器中的生物学功能乃至病毒的复制机制发挥重要作用。本研究中发现,M蛋白N端或C端与APEX2融合表达后并未改变其定位至线粒体的能力,暗示APEX2标签的引入极有可能并未改变M蛋白定位线粒体并由此引发的线粒体凋亡效应,这为进一步开展基于APEX2标记技术解析RABV病毒M蛋白诱导线粒体凋亡途径的具体分子细节奠定了基础。
利益冲突:无
猜你喜欢
华人时刊(2022年9期)2022-09-06 01:02:44
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
华人时刊(2020年15期)2020-12-14 08:10:36
动漫星空(兴趣百科)(2020年3期)2020-03-24 02:33:32
湖南畜牧兽医(2016年3期)2016-06-05 08:37:55
兽医导刊(2016年12期)2016-05-17 03:51:15
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
中国卫生产业(2015年10期)2015-03-11 18:58:41
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
