
以不同碳链长度脂肪酸甘油三酯为油相的微乳液特性研究
2023-07-29 10:19付兆燕邱斌刘玮刘霞齐沙沙尤艳莉
食品工业 2023年7期
付兆燕 ,邱斌,刘玮,刘霞,齐沙沙,尤艳莉
1.烟台大学(烟台 264010);2.山东省农业科学院农产品加工与营养研究所(济南 250000)
微乳液(microemusion,Me)是由油相、表面活性剂、助表面活性剂和水相组成的外观澄清透明,各向同性的热力学稳定体系。微乳液通常有3种类型:水包油型微乳液,常作为许多脂溶性活性成分的输送载体[1-2];油包水型微乳液,适用于负载水溶性活性成分;双连续型微乳液同时具有亲脂性和亲水性[3]。微乳液具有热力学稳定、可自发形成、表面张力低、粒径小和黏度低等特点,被广泛应用在化学、生物、医学及食品领域[4]。
不同油相在性质、功能和应用等方面具有不同的特点。中链甘油三酯(MCT)在体内的代谢是通过门静脉直接运输到体循环[5],能快速供能,且MCT流动性高、自乳化能力强,因此MCT是作为微乳液油相的首选[6-7]。花生油、核桃油和亚麻籽油等长链甘油三酯(LCT),含有丰富的多不饱和脂肪酸(PUFA),具有抗炎、抗肿瘤、保护心血管等作用[8],但PUFA容易氧化,又因碳链过长不易制备成微乳液,因此本文将MCT与LCT复配作为微乳液的油相,既可以提高MCT和LCT的水溶性,也可以保留不同脂肪酸的功能特性,提高其生物利用度。为微乳液在食品工业领域的广泛应用提供参考。
1 材料与方法
1.1 材料与试剂
高油酸花生油、有机山核桃油、亚麻籽油(市售);吐温80、无水乙醇、丙三醇、亚甲基蓝、乙醚、氯化钠(均为分析纯,国药集团);苏丹红Ⅰ(美仑生物技术有限公司);去离子水(实验室自制)。
1.2 仪器与设备
AY220电子分析天平(德国赛多利斯仪器有限公司);79-1磁力搅拌器(鸿科仪器厂);SYNERGY HTX酶标仪(美国Bio Tek仪器有限公司);Zetasizer Nano S纳米粒度分析仪(马尔文仪器有限公司);HR20流变仪、示差量热扫描仪(TA Instruments-Waters LLC)。
1.3 方法
1.3.1 不同油相微乳液的制备
参照Garti等[9]的方法,将油相与助表面活性剂(乙醇)混合形成混合油相(1︰3,W/W),水相与助表面活性剂(丙三醇)混合形成混合水相(1︰1,W/W),混合均匀后取混合油相与表面活性剂混合,逐滴加入混合水相至体系的90%(质量比)。
油相选择亚麻籽油、高油酸花生油、山核桃油3种含有不同长链脂肪酸的食用油与MCT复配,复配比例为3︰2(W/W);表面活性剂为吐温80;助表面活性剂为乙醇及丙三醇。亚麻籽油-MCT微乳液为Me-1,高油酸花生油-MCT微乳液为Me-2,山核桃油-MCT微乳液为Me-3。
1.3.2 微乳液的类型鉴别
试验采用染色法进行微乳液类型鉴别[10]。将少量苏丹红和亚甲基蓝分别溶于1 mL乙醚和水中。取20 μL染色液加入装有微乳液的比色管中,观察染料在微乳液中的扩散情况。若亚甲基蓝扩散速度大于苏丹红,则微乳液为O/W型;反之,则微乳液为W/O型。
1.3.3 微乳液的结构表征
1.3.3.1 透射电镜
微乳液的微观形态通过透射电子显微镜进行检测。取少量微乳液滴加到覆盖在300目(48 μm)铜网的支撑膜上,自然干燥10 min。观察前,滴加2%(质量分数)磷钨酸染色剂染色15 min,自然干燥10 min,用透射电子显微镜进行观察和拍摄,加速电压为75 kV。
1.3.3.2 粒径大小及分布
微乳液的粒径分布和多分散性采用马尔文激光粒度仪纳米ZS光散射仪,通过动态光散射技术进行测量。
1.3.4 微乳液的稳定性研究
1.3.4.1 离心稳定性
取适量制备好的微乳液,按2 000,4 000,8 000和12 000 r/min离心30 min,以去离子水为空白对照,测定离心前后波长550 nm下的吸光度。透光率按式(1)计算。
式中:T为透光率,%;A0为离心前微乳吸光度;A1为离心后微乳吸光度。
1.3.4.2 盐度稳定性
分别配制0.1,0.5和1.0 mol/L的盐溶液作为水相制备微乳液,以去离子水制备的微乳液为条件对照,在波长550 nm下测定其吸光度,考察不同盐度对微乳液稳定性的影响。透光率按式(2)计算。
式中:T为透光率,%;A2为纯水制备微乳吸光度;A3
为不同盐度的微乳吸光度。
1.3.4.3 温度稳定性
将微乳液在-18,4,25,37和60 ℃储存5 h,恢复室温后,在波长550 nm下测定其吸光度,以室温下的微乳液为条件对照,考察不同温度对微乳液稳定性的影响。透光率按式(3)计算。
式中:T为透光率,%;A4为室温下微乳吸光度;A5为不同温度储存后的微乳吸光度。
1.3.5 微乳液的流变性能分析
采用流变仪,对1.3.1中制备的亚麻籽油-MCT微乳液(Me-1)、花生油-MCT微乳液(Me-2)、山核桃油-MCT微乳液(Me-3)进行流变性能测试。
1.3.5.1 稳态流动扫描测试
流动扫描测试是通过施加一个剪应力斜坡,并等待得到的剪切速率随时间的斜率在每个点小于0.001%时,可以假定几乎达到稳态[11]。测试所用平行板直径40 mm,微乳液与锥板的间隙500 μm,剪切速率0.1~100 s-1,温度25 ℃;温度扫描测试的剪切速率50 s-1,温度区间5~70 ℃。
1.3.5.2 动态振荡扫描测试
在固定振荡频率下进行应变扫描测试,应变扫描中所用平行板直径40 mm,微乳液与锥板间隙500 μm,频率1 Hz,温度25 ℃,应变范围0.1%~100%;频率扫描测试的应变值2%,频率范围0.1~100 rad/s。
1.3.6 微乳液的热分析
采用差示扫描量热仪(DSC),对3种不同油相的微乳液体系中各单组分和混合水相(去离子水-丙三醇1︰1,V/V),以及不同含水量的微乳液进行热行为分析。测试方法:取适量样品置于铝盘中,称量后密封处理,将密封后的铝盘放入DSC进样盘中。测试条件:初始温度为25 ℃,以25 ℃/min的速度降到-80 ℃,恒温5 min后以5 ℃的速度升温50 ℃到结束检测。
1.3.7 数据处理
采用Origin 2021b软件绘制图表,使用SPSS 22.0进行数据统计分析,P<0.05认为有统计学差异。每个样品至少进行3次重复试验,试验数据以平均值±标准偏差表示。
2 结果与讨论
2.1 不同油相微乳液的制备
通过1.3.1的方法制备得到的以中/长碳链脂肪酸复配为油相的微乳液,均为外观澄清透明的体系,如图1所示。

图1 微乳液的外观图
2.2 类型鉴别分析
从图2可以看到,在3种微乳液中亚甲基染料的扩散速度均快于苏丹红染料,因此Me-1,Me-2和Me-3均为O/W型微乳液。

图2 染色法外观图
2.3 结构表征
2.3.1 透射电镜
透射电镜(TEM)是表征微乳液微观结构的手段之一[12]。从图3可以看到3种微乳液液滴均为球形,大小均一,粒径均小于100 nm,具体粒径大小需要与纳米激光粒度仪测试结果相结合。

图3 不同油相微乳液透射电镜图
2.3.2 粒径大小及分布
动态光散射技术(DLS)是一种快速简单的测量微乳液粒径大小及其分布的方法[13],也是常用的表征物质结构的手段之一。其原理是粒子进行布朗运动的速度与粒径大小有关,粒径越小,布朗运动的速度越快,散射光照射在粒子上时,布朗运动的速度影响光信号的强弱,基于这一点DSL技术可以有效测量微乳液粒径[14]。如表1所示,3种不同油相制备的微乳液平均粒径均小于100 nm,符合微乳液粒径要求,说明体系为单分散体系。

表1 微乳液粒径及其分布
2.4 稳定性分析
2.4.1 离心稳定性
微乳液是一个热力学稳定的体系,将不同碳链长度脂肪酸复配为油相的3种微乳液在不同转速下离心30 min后,均未出现分层现象,外观澄清透明。在图4中可以看到,Me-1和Me-3在高速离心后的透光率略小于低速离心后的透光率,原因可能是随着转速的增大微乳液部分颗粒出现聚集,使得透光率下降,但总体透光率均大于95%,表明3种微乳液具有良好的离心稳定性。

图4 离心稳定性
2.4.2 盐度稳定性
微乳液中无机盐的含量,对微乳液相行为有很大影响[15],从图5可以看到,3种不同油相的微乳液随着盐度的增加,透光性出现下降趋势,原因可能在于表面活性剂的亲水端与水相相连产生氢键,从而提高微乳液的稳定性,盐离子的加入会使表面活性剂的极性部分减少,进而影响氢键的形成。Kim等[16]在茶树精油微乳液的稳定性研究中,也得到类似结论。总体各微乳液的透光率都在95%以上,具有良好耐盐性能。
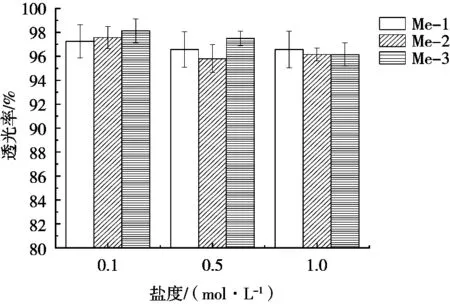
图5 盐度稳定性
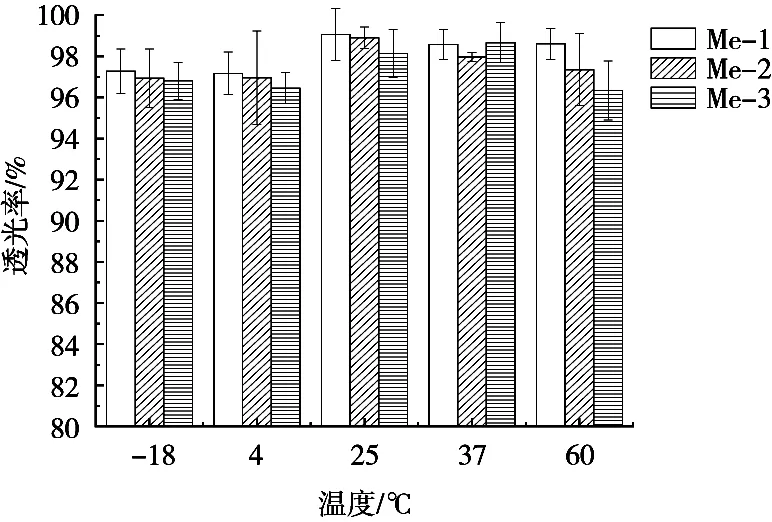
图6 温度稳定性
2.4.3 温度稳定性
将微乳液在不同温度下放置5 h,恢复室温后,体系未发生分层,仍呈澄清透明状态。不同油相的3种微乳液,在低温(-18和4 ℃)条件下,透光率比在常温(25和37 ℃)要低,表明低温对表面活性剂亲水端与水相的结合产生影响,而高温(60 ℃)对透光率的影响并不明显。整体透光率均在95%以上,说明3种微乳液都具有良好的热稳定性。
2.5 流变性能分析
2.5.1 稳态流动扫描测试
图7展示25 ℃下,3种不同油相制备的微乳液表观黏度(η)随剪切速率(γ)的关系及剪切应力(σ)随剪切速率(γ)的关系。因为小于1 s-1的剪切速率接近扭矩下限,因此此区间的表观黏度均表现出一定范围波动[17],而剪切速率大于1 s-1时,微乳液则表现出线性的、稳定的流动曲线,即体系为牛顿流体。与此同时,在剪切应力与剪切速率的关系中Me-1,Me-2和Me-3均呈线性上升趋势,符合在牛顿流体中,剪切应力不依赖于剪切速率的特点。该结果与Abbasi等[2]制备的菜籽油-卵磷脂-正丙醇微乳液及Pessoa等[18]制备的巴巴苏油微乳液的研究结论相符。
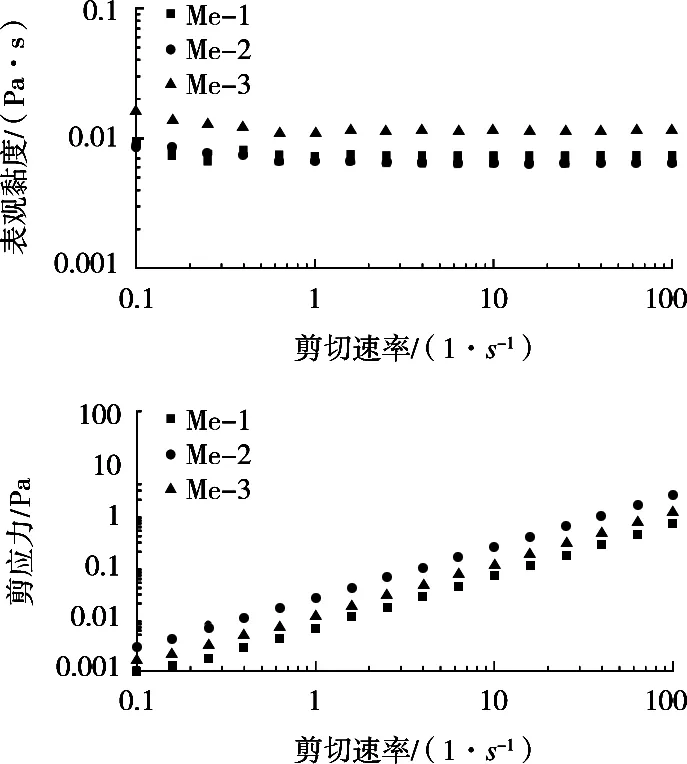
图7 不同油相的微乳液稳态流动曲线
牛顿流体的模型方程如式(4)所示。
式中:σ为剪切应力,η为表观黏度,γ为剪切速率。
对图7的流动曲线进行拟合,得到各微乳液体系的表观黏度,结果如表2所示。

表2 不同油相微乳液的表观黏度
微乳液体系温度触变性如图8所示。3种微乳液均表现出表观黏度不随剪切时间的增长而变化的特点。说明微乳液体系不产生微结构,其黏度不受剪切历史的影响,无明显剪切触变性。

图8 25 ℃下恒剪切速率(50 s-1)表观黏度与剪切时间变化的关系
2.5.2 动态扫描测试
通过对微乳液进行动态扫描可以得到体系的黏弹性等流变特性。试验将不同油相的微乳液进行应变扫描,确定线性黏弹区,如图9(A)所示。在应变值大于1%后的黏性模量(G’)不依赖于应变,因此将应变值设置为2%。如图9(B)所示,在频率扫描中各微乳液体系均表现出与稀溶液相似的黏性行为,且黏性模量(G’’)值较低。在整个扫描范围内,黏性模量与频率有明显依赖关系。
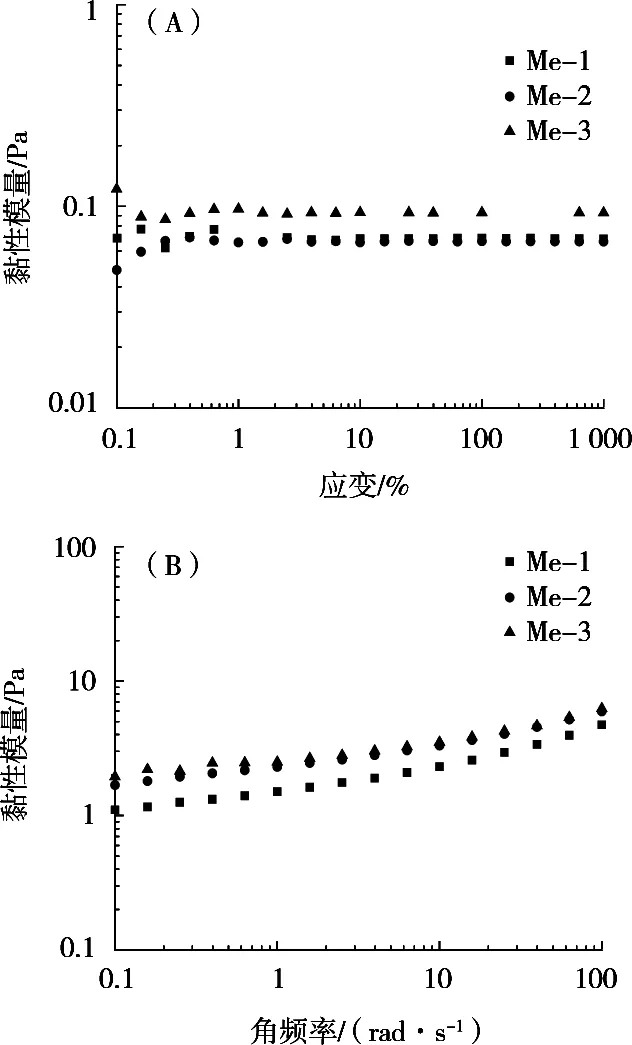
图9 不同油相微乳液应变扫描(A)及不同油相微乳液频率扫描(B)
2.6 微乳液的热分析
DSC是在程序控制温度下,测量进出样品和参比物之间的热流差或功率差与温度或时间关系的一种技术[19]。为了解多组分共存的微乳液体系,试验对微乳液单一组分进行DSC扫描。如表3所示,去离子水在0.36 ℃附近一个吸热峰,与水的熔点理论值0 ℃相似。在吐温80的DSC扫描中,升温过程中出现2个明显的吸放热峰,这可能与吐温80分子结构有关,在降温过程中,分子难以形成规则的结构,而升温过程中,分子的活动上升,分子重新排列为规则结构,表现为冷结晶放热,温度升高至其熔点,开始吸热熔融[20]。由文献得知,乙醇的熔点为-114 ℃,不在DSC扫描范围内,因此未对其进行DSC扫描[21]。丙三醇在整个扫描过程中没有出现任何吸热或放热峰。
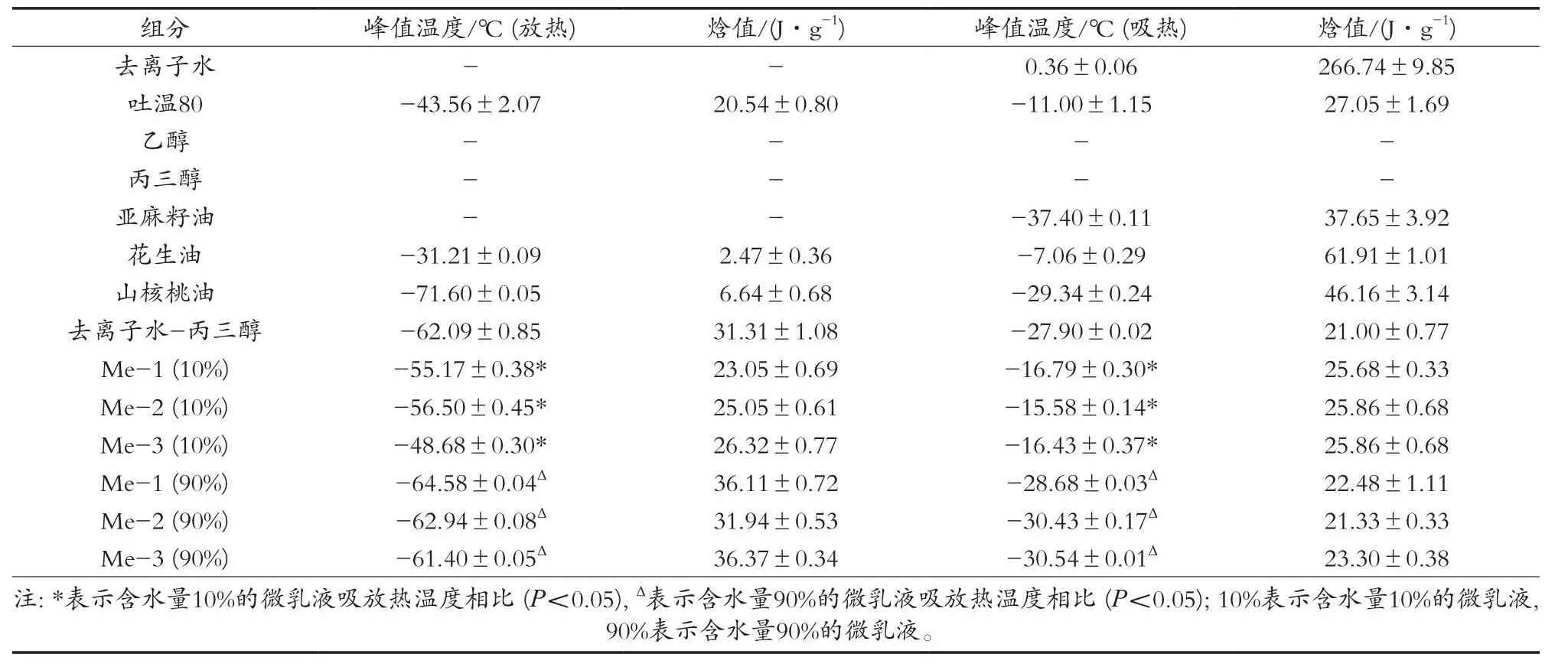
表3 微乳液体系各组分DSC扫描数据
亚麻籽油和山核桃油分别在-37.40和-29.34 ℃附近出现熔融峰,花生油在-7.06 ℃附近出现熔融峰。这可能与3种食用油中脂肪酸组成有关,花生油含有大量单不饱和脂肪酸,氧化稳定性较好,而亚麻籽油和山核桃油含有丰富的多不饱和脂肪酸,容易发生氧化[22]。
混合水相(Water-Gly=1︰1)相比水的吸热峰从0.36 ℃降低到-27.90 ℃,焓值从266 J/g降低至21 J/g。混合水相晗值减小的原因,一是丙三醇成为体系中水相的一部分[24],二是有一部分的水与丙三醇之间相互作用产生氢键[25]。
如图10所示,通过DSC扫描发现,Me-1a,Me-2a和Me-3a这3种含水量10%的微乳液在升温曲线的前半段的吸、放热峰与吐温80的出峰相似,此时体系中表面活性剂占比最大,因此可将其看作是体系中表面活性剂的热行为。Me-1b,Me-2b和Me-3b为含水量90%的微乳液,3种微乳液的出峰时间和焓值与混合水相的扫描结果相似,此时体系中水相作为连续相,即水包油型微乳液,因此图谱中只显示水相相关的出峰。
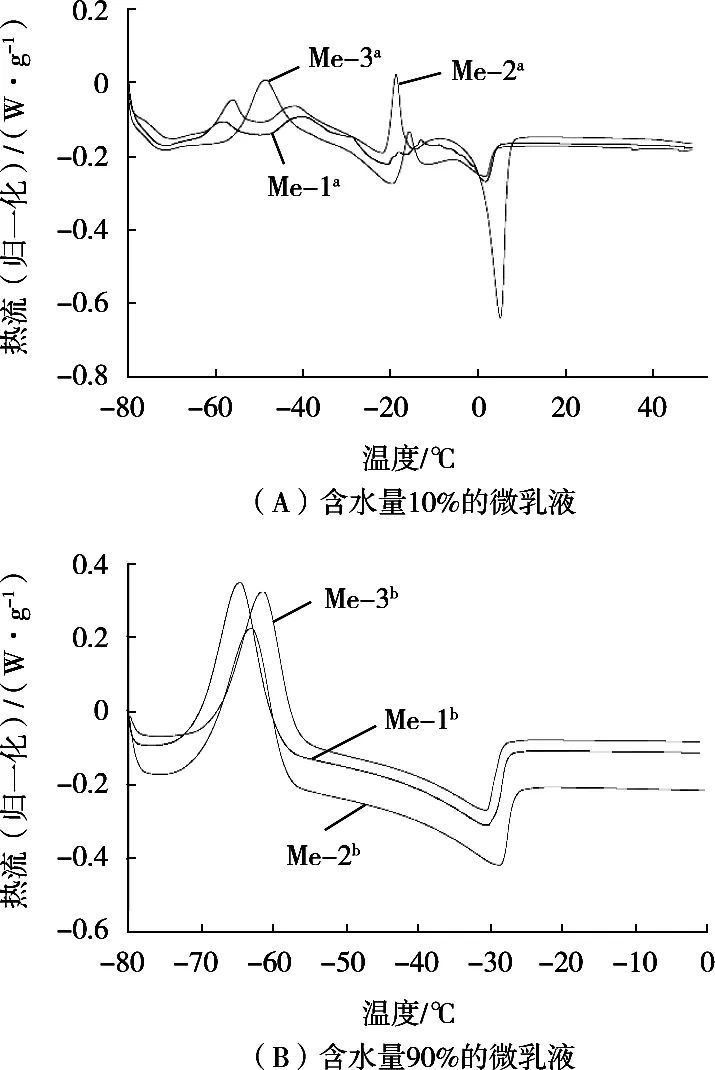
图10 不同油相微乳液DSC扫描曲线
对含水量10%和90%的3种微乳液的吸放热峰值温度及对应焓值分别进行方差分析,研究不同碳链的油相对微乳液的热力学影响(表3),结果表明,含水量10%和90%的微乳液温度变化无明显规律,但含水量不同的微乳液均表现出不同碳链长度的油相对微乳液体系熔融和结晶温度的影响存在显著性差异(P<0.05)。
3 结论
以亚麻籽油、花生油、山核桃油分别与MCT复配为油相(3︰2,W/W),吐温80为表面活性剂,乙醇和丙三醇为助表面活性剂制备的3种微乳液体系(混合油相-表面活性剂-混合水相1︰9︰90,W/W),外观均澄清透明,微观结构呈球型,平均粒径分别为48.97,30.92和10.51 nm,多分散指数为0.14,0.10和0.01,属于水包油型微乳液。稳定性研究显示,3种体系在不同的离心、盐度、温度条件下,透光率均大于95%,证明3种微乳液有较强的离心稳定性,耐盐性及耐热性。流变学研究显示,3种微乳液均为弹性较小牛顿流体,无剪切触变性。热力学研究显示,助表面活性剂丙三醇的加入使得体系中水相的焓值降低,不同碳链的油相对微乳液的热力学影响存在显著性差异(P<0.05)。试验可为制备高稳定性的复合油相微乳液提供新思路。
猜你喜欢
河北果树(2022年1期)2022-02-16
北京农学院学报(2021年4期)2021-11-09
煤矿爆破(2020年3期)2020-12-08
石油地质与工程(2019年3期)2019-09-10
发光学报(2019年8期)2019-08-20
中国钼业(2019年2期)2019-01-19
中国洗涤用品工业(2017年2期)2017-04-16
水利技术监督(2016年6期)2017-01-15
浙江大学学报(工学版)(2016年11期)2016-06-05
遵义医科大学学报(2016年1期)2016-03-16
