
抑制NLRP3/IL-1β信号通路对高尿酸血症CKD大鼠肾功能的影响*
2023-07-07 09:14:50曹文琼黄新梅高红梅朱学新马潇贾新燕
西部医学 2023年6期
曹文琼 黄新梅 高红梅 朱学新 马潇 贾新燕
(兰州市第一人民医院肾病科,甘肃 兰州 730050)
近年来,慢性肾脏病(Chronic kidney disease,CKD)的发病率不断增加。CKD是一种多因素疾病,糖尿病和高血压是导致CKD的主要原因,此外,还有一些其他常见原因,包括肾小球肾炎、多囊肾病、肾结石、尿路感染和肾毒素等[1]。CKD的确切机制尚不清楚,最终共同途径涉及肾小球硬化、血管硬化和肾小管间质纤维化。高尿酸血症是导致肾功能损害的重要因素,研究[2]表明,高尿酸血症可通过多种机制加重CKD患者的肾功能损害,包括直接毒性作用、促进炎症反应、激活肾素-血管紧张素-醛固酮系统、诱导线粒体功能障碍以及氧化应激等。据统计,我国CKD患者高尿酸血症的患病率为36.6%~50%[3-4],且随着CKD的进展呈现不断增加的趋势。
NOD样受体家族核苷酸结合寡聚化结构域样受体3(NOD-like receptor family,pyrin domain containing 3,NLRP3) 属于核苷酸结合寡聚化结构域样受体家族成员,作为固有免疫系统的模式识别受体,可识别病原体相关分子模式以及损伤相关分子模式。活化的NLRP3能够与凋亡相关斑点样蛋白和pro-Caspase-1相互作用,形成胞质复合物从而产生具有活性的Caspase-1,并触发下游促炎介质IL-1β的成熟和释放,从而参与各种炎症性疾病[5-6]。已有研究[7]表明,高尿酸血症肾病患者外周血单核细胞中NLRP3和Caspase-1的mRNA表达水平明显升高,血浆内IL-1β、TNF-α、IL-6、IL-18水平也均升高,因此推测NLRP3/IL-1β信号通路及其相关炎症因子参与了高尿酸所致CKD的肾功能损害。此外,在缺血/再灌注诱导急性肾损伤后引起的CKD 模型小鼠中检测到血清、尿液和肾组织中NLRP3表达均升高,并检测到肾组织内NLRP3信号通路相关炎症因子表达上调,这一现象与肾功能损害程度密切相关[8]。鉴于上述内容,为了验证干预高尿酸血症CKD中NLRP3/IL-1β信号通路能否对肾功能起到保护作用,本研究通过构建高尿酸血症CKD大鼠模型并以NLRP3抑制剂MCC950与IL-1β抑制剂GIBH-130进行作用, 观察该作用下大鼠模型相关指标变化,并初步探讨作用机制。
1 资料与方法
1.1 实验动物 体重为180~220 g的雄性SD大鼠,由甘肃中医药大学提供,健康且无特定病原体,饲养条件设置为温度23~25℃,相对湿度控制在55%~65%,12 h/12 h交替照明,噪音小于60分贝,环境经严格灭菌处理,环境清洁、通风良好。本研究通过医院伦理委员会审核。
1.2 主要试剂 氧嗪酸钾购于美国Sigma公司;NLRP3抑制剂MCC950购于美国MedChemExpress公司;IL-1β抑制剂GIBH-130和免疫组织化学染色(DAB显色)试剂盒购于北京百奥莱博科技有限公司;IL-1β、TNF-α、IL-18 ELISA测定试剂盒购于江苏凯基生物公司;SOD与MDA检测试剂盒购于上海钰博生物科技有限公司;HE染色试剂盒和PAS染色试剂盒购于北京索莱宝生物公司;Epon812、醋酸铀和柠檬铅购于美国EMS公司;PVDF膜购于美国Millipore公司;BCA蛋白测定试剂盒、DAB显色试剂盒和ECL增强化学发光液购于上海碧云天生物公司;兔抗人Nephrin、Podocin、NLRP3、IL-1β、Caspase-1、GAPDH多克隆抗体以及辣根过氧化物酶标记的山羊抗兔IgG等抗体均购于英国Abcam公司。
1.3 方法
1.3.1 动物造模 参考文献方法[9]制备高尿酸血症CKD大鼠模型。将30只SD大鼠适应性饲养1周后,切除右肾,按照数字随机表法分为模型组、NLRP3抑制剂组、IL-1β抑制剂组,每组10只。术后1周,3组大鼠均开始以800 mg/kg的剂量灌胃氧嗪酸钾,每日一次,连续24 d,以构建高尿酸血症CKD模型。同时,NLRP3抑制剂组大鼠以10 mg/kg 的剂量灌胃NLRP3抑制剂MCC950,1次/日,连续24 d;IL-1β抑制剂组大鼠以0.25 mg/kg的剂量灌胃IL-1β抑制剂GIBH-130,1次/日,连续24 d;另取10只健康SD大鼠作为对照组,以等容量蒸馏水灌胃。 实验期间,所有大鼠均提供普通饲料和蒸馏水以自由饮食饮水。
1.3.2 血清生化指标 测定各组大鼠在最后一次给药24 h后,通过眶静脉采血,室温静置2 h后,以3500 rpm/min 离心10 min,收集上清。通过全自动生化分析仪测定血清中SUA、BUN和Scr水平,具体根据检测试剂盒说明书进行测定。
1.3.3 肾组织炎症因子水平检测 各组大鼠取血后处死,在无菌环境下解剖取左肾组织,剪取约0.5 g肾组织,加入预冷的PBS缓冲液,研磨制成匀浆,以4000 rpm/min离心10 min,收集上清液,BCA法测定蛋白浓度,采用ELISA法检测上清液中IL-1β、TNF-α及IL-18水平,按照试剂盒说明书进行操作,制作标准曲线并计算各因子含量。
1.3.4 HE染色 将解剖的各组大鼠左肾组织固定在10%中性福尔马林液中,24 h后经梯度酒精脱水,常规石蜡包埋,切片机上连续切成厚度为4 μm的组织薄片。将切片脱蜡至水,置入苏木精染色5 min,流水冲洗后,再于伊红染色液中染色3 min,流水再次冲洗,脱水后晾干,透明,中性树胶封片,通过光学显微镜观察肾组织形态变化,摄取图像。
1.3.5 PAS染色 各组大鼠肾组织切片经脱蜡至水后,置入阿利新蓝溶液中染色10 min,流水冲洗,利用过碘酸溶液氧化5 min,再浸入SchiffReagent试剂液中染色10 min,流水冲洗,苏木素染色液复染,酸性乙醇分化,氨水返蓝,流水冲洗干净,脱水与透明,中性树胶封片,通过光学显微镜观察肾组织染色情况,摄取图像。
1.3.6 透射电子显微镜 观察各组大鼠肾组织在2.5%戊二醛中固定,切片机上将组织切成大小约为1 mm3的组织块。固定好后,丙酮脱水,Epon812浸透,纯包埋液完全包埋,通过超薄切片机切成厚度约为1 μm的组织薄片,经醋酸铀和柠檬酸铝染色后,通过透射电镜观察足细胞超微结构变化,摄取图像。
1.3.7 免疫组织化学染色 将固定好的肾组织进行常规石蜡包埋,切片机切成4 μm 的石蜡组织切片。切片进行脱蜡处理,滴加柠檬酸缓冲液高温高压抗原修复,用3%H2O2室温处理30 min。滴入山羊血清室温封闭30 min,然后滴加稀释的兔抗人Nephrin多克隆抗体(1∶100)和兔抗人Podocin多克隆抗体(1∶100),置于4℃孵育过夜;次日,PBS 冲洗,滴加对应稀释的二抗(1∶1000),37℃孵育30 min。PBS冲洗,利用DAB显色,流水冲洗,在苏木精溶液中复染,经脱水与透明后,晾干,中性树胶封片,通过光学显微镜观察组织内着色情况,摄取图像,胞质、胞核染为棕色至棕褐色为阳性,随机选择5个视野,通过ImageJ软件进行分析,分别计算Nephrin和Podocin阳性表达率。
1.3.8 Western blot法 将各组大鼠肾组织在无菌环境下剪碎,加入预冷蛋白裂解液裂解,收集上清,BCA法定量检测蛋白浓度。取等量各组蛋白样品分别与5×Loading buffer混合,制备10% SDS-PAGE凝胶,将蛋白上样至凝胶孔内,恒压电泳分离蛋白质,并电转移至PVDF膜,5%脱脂奶粉室温封闭2 h,将稀释的兔抗人NLRP3、IL-1β、Caspase-1多克隆抗体(1∶1000)作为一抗,与膜共置于4℃下孵育过夜;次日,TBST洗膜,加入辣根过氧化物酶标记的山羊抗兔IgG(1∶5000)作为热炕,与膜室温孵育1 h,TBST再次洗膜,ECL试剂液显影,X射线胶片曝光后定影,Image J图像分析软件分析条带灰度值,内参蛋白为GAPDH,以目的蛋白与GAPDH的灰度值之比作为NLRP3、IL-1β以及Caspase-1的蛋白相对表达水平。
1.3.9 氧化应激指标测定 取各组大鼠左肾组织,无菌环境下剪碎并加入裂解液,利用匀浆机制备匀浆液,以4000 rpm/min离心20 min,收集上清液,检测SOD活性与MDA含量,步骤参照各试剂盒说明书进行。

2 结果
2.1 各组大鼠血清肾功能指标 通过检测各组大鼠血清SUA、BUN和Scr来评价肾功能,与对照组比较,模型组大鼠血清SUA、BUN和Scr水平均显著升高(P<0.05);与模型组比较,NLRP3抑制剂组和IL-1β抑制剂组大鼠血清SUA、BUN以及Scr水平均显著降低(P<0.05)。见表1。
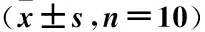
表1 各组大鼠血清SUA、BUN和Scr水平比较Table 1 Comparison of serum SUA, BUN and Scr levels of rats in each group
2.2 各组大鼠炎症因子水平比较 ELISA检测各组大鼠肾组织炎症因子含量的结果显示,与对照组比较,模型组大鼠肾组织IL-1β、TNF-α和IL-18含量均显著升高(P<0.05);而NLRP3抑制剂组大鼠肾组织IL-1β、TNF-α和IL-18含量均显著低于模型组(P<0.05);IL-1β抑制剂组大鼠肾组织IL-1β、TNF-α和IL-18含量也均显著低于模型组(P<0.05)。见表2。
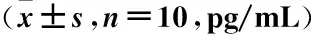
表2 各组大鼠肾组织IL-1β、TNF-α和IL-18含量比较Table 2 Comparison of IL-1β, TNF-α and IL-18 contents in kidney tissue of rats in each group
2.3 各组大鼠肾组织病变观察 经过HE染色和PAS染色观察到,对照组大鼠肾组织形态清晰,肾小球、肾小管及肾间质均未见明显病理改变;模型组大鼠肾组织结构异常,可见肾小管上皮细胞明显坏死、萎缩,肾小球系膜增生,系膜细胞减少,并有较多炎性细胞浸润等损伤现象;而相较于模型组,NLRP3抑制剂组和IL-1β抑制剂组大鼠肾组织病理损伤均减轻,形态结构有所恢复。见图1。

图1 各组大鼠肾组织病理学形态观察(上HE,100×;下PAS,100×)Figure 1 Histopathological observation of rat kidney in each group
同时,透射电镜下可见对照组大鼠肾小球足细胞结构清晰,绝大多数足突排列整齐,贴附于肾小球基底膜外侧;模型组大鼠肾小球足细胞足突增粗,出现融合或消失的现象,且肾小球基底膜增厚;相较于模型组,NLRP3抑制剂组和IL-1β抑制剂组大鼠足细胞足突融合或消失现象得到改善,肾小球基底膜也未见明显增厚。见图2。
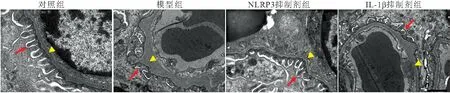
图2 各组大鼠肾组织足细胞超微结构变化(透射电镜,20000×)Figure 2 Ultrastructural changes of podocytes in renal tissue of rats in each group 注:红色箭头为足突,黄色为基底膜。
2.4 各组大鼠肾组织Nephrin与Podocin表达比较 免疫组织化学染色检测结果显示,对照组大鼠肾组织内可见明显阳性染色,模型组大鼠肾组织中阳性染色较对照组变浅,Nephrin阳性率与Podocin阳性率均显著减少(P<0.05);与模型组比较,NLRP3抑制剂组和IL-1β抑制剂组大鼠肾组织中阳性染色均加深,Nephrin阳性率与Podocin阳性率也均显示为显著增加(P<0.05)。见图3。
2.5 各组大鼠肾组织NLRP3/IL-1β信号通路蛋白水平比较 Western blot检测结果显示,与对照组比较,模型组大鼠肾组织内NLRP3、IL-1β及Caspase-1蛋白相对表达量均显著上调(P<0.05);与模型组比较,NLRP3抑制剂组和IL-1β抑制剂组大鼠肾组织内NLRP3、IL-1β、Caspase-1蛋白相对表达量均显著下调(P<0.05)。见图4。

图4 Western blot检测各组大鼠肾组织NLRP3/IL-1β信号通路蛋白表达Figure 4 Western blot detection of NLRP3/IL-1β signaling pathway protein expression in kidney tissue of rats in each group注:1.对照组;2.模型组;3.NLRP3抑制剂组;4.IL-1β抑制剂组。与对照组比较,①P<0.05;与模型组比较,②P<0.05。
2.6 各组大鼠肾组织氧化应激指标比较 经过检测各组大鼠肾组织SOD活性和MDA含量发现,模型组大鼠肾组织SOD活性显著低于对照组,而MDA含量显著高于对照组(P<0.05);与模型组相比,NLRP3抑制剂组和IL-1β抑制剂组肾组织SOD活性均显著升高,MDA含量均显著减少(P<0.05)。见图5。

图5 各组大鼠肾组织SOD和MDA水平检测Figure 5 Detection of SOD and MDA levels in kidney tissue of rats in each group注:与对照组比较,①P<0.05;与模型组比较,②P<0.05。
3 讨论
高尿酸血症是高血压、痛风性关节炎、代谢综合征以及CKD发生的关键危险因素。肝脏尿酸生成过多和肾脏排泄不足均可能导致高尿酸血症,尿酸是嘌呤分解代谢的最终氧化产物,在人体中其浓度的改变通常与肾脏中尿酸结晶的形成有关,从而导致尿酸结石,引起强烈的炎症反应并引起肾脏损伤[10]。目前,随着人类生活条件和生活方式的改善,高尿酸血症肾病的发病率在世界范围内迅速增加。针对高尿酸血症CKD的治疗策略主要集中在使用黄嘌呤氧化还原酶抑制剂减少体内尿酸的产生或者利用促尿酸排泄剂促进尿液中尿酸的排泄。然而,这两种治疗剂均由于引起的不良副作用而使得临床使用受限,例如,别嘌呤醇是常用的黄嘌呤氧化还原酶抑制剂,可损害嘧啶代谢导致部分使用者出现严重的超敏反应和粒细胞缺乏症,并进一步加重了肾毒性和肾损伤[11]。因此,迫切需要寻找有效且毒性小的药物来治疗高尿酸血症CKD。
NLRP3炎症小体由模式识别受体蛋白NLRP3、包含CARD的凋亡相关斑点样蛋白和效应蛋白前Caspase-1组成。作为NLR炎性通路中的重要组成部分,可被细菌、病毒等多种病原体危险信号刺激后活化,被激活的NLRP3能够募集凋亡相关斑点样蛋白与前体Caspase-1形成炎症小体复合物,并通过自我切割产生具有活性的Caspase-1;Caspase-1作为IL-1β转化酶,经自身催化激活后,可将无活性的促炎因子IL-1β和前体IL-18的前体剪切加工为成熟的IL-1β和IL-18并释放出来,进一步增加局部炎性相关基因表达,从而诱导炎症反应和组织损伤[6],此外,Caspase-1还能够启动细胞死亡程序[12]。研究[13-14]表明,NLRP3/IL-1β信号通路参与多种疾病的发生发展,包括自身免疫性疾病、感染和非感染性疾病。同样,该通路参与肾脏疾病的炎症反应,可引起病理性病变和肾脏损伤,在肾脏疾病进程中起着重要作用。例如,Zhao等[15]研究表明NLRP3炎症小体的激活可通过诱导线粒体功能障碍来调节Ang II诱导的足细胞损伤,抑制或阻断NLRP3可明显减轻线粒体功能障碍引起的肾损伤;Krishnan等[16]研究发现在高血压小鼠模型中,使用NLRP3抑制剂MCC950可以显著降低血压和肾组织纤维化,抑制肾脏中IL-18和IL-1β的表达,并减轻了肾功能障碍。同样,本研究表明,经NLRP3抑制剂MCC950和IL-1β抑制剂GIBH-130作用的高尿酸血症CKD大鼠肾组织内IL-1β、TNF-α和IL-18含量均下降,这提示肾组织炎症反应减轻。高尿酸血症加重肾脏负担,造成肾脏损害,引起肾脏损害相关指标如Scr 和BUN升高,经NLRP3抑制剂MCC950和IL-1β抑制剂GIBH-130作用也明显降低了血清SU A、BUN和Scr水平,因而改善了肾功能。此外,组织病理学观察结果还表明,NLRP3抑制剂MCC950和IL-1β抑制剂GIBH-130治疗均明显减轻了高尿酸血症CKD大鼠的肾损伤,提高了Nephrin和Podocin的表达水平。Nephrin是由足细胞表达的位于裂孔隔膜上的跨膜蛋白,能够与Podocin结合形成复合体以维持裂孔隔膜的结构完整,这两种特异性蛋白是肾小球基底膜的关键成分[17]。以上这些结果均表明,利用抑制剂靶向抑制NLRP3/IL-1β信号通路可显著改善高尿酸血症CKD大鼠肾功能。
氧化应激是在各种因素刺激下机体活性氧或自由基相对超负荷,引起细胞内多种生物大分子发生损伤,从而对生命活动产生影响的应激反应。肾脏组织是一个高度代谢的器官,线粒体中富含氧化反应,这使得其容易受到氧化应激造成的损害[18]。氧化应激可以加速肾脏疾病的进展,此外,在CKD晚期患者中,氧化应激增加与高血压、动脉粥样硬化、炎症和贫血等并发症有关[19]。有研究[20]表明,线粒体能够通过释放活性氧促进DNA释放到胞质溶液中,激活NLRP3炎症小体并引起肾脏疾病,可见氧化应激在肾脏疾病的发生中起着重要作用,而通过抑制氧化应激来调节NLRP3途径活化,可达到改善肾功能的作用[21]。本研究显示,高尿酸血症CKD大鼠肾组织SOD活性下降而MDA含量升高,表明组织内发生了氧化应激反应;在NLRP3抑制剂MCC950和IL-1β抑制剂GIBH-130作用下,大鼠肾组织SOD活性升高,同时MDA含量下降,这提示组织内氧化应激受到抑制。
4 结论
NLRP3/IL-1β信号通路与高尿酸血症CKD发生发展有关,通过抑制该途径可阻止肾组织炎症因子释放,减轻肾组织损伤,并抑制氧化应激反应,从而起到对肾功能的保护作用。由此推测,NLRP3/IL-1β信号通路可作为高尿酸血症CKD治疗的潜在靶点。
猜你喜欢
中老年保健(2021年4期)2021-08-22 07:07:36
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:05:56
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
家庭医学(下半月)(2019年9期)2019-10-12 08:03:56
中医眼耳鼻喉杂志(2019年2期)2019-04-13 05:23:46
西南军医(2016年6期)2016-01-23 02:21:19
西南军医(2015年2期)2015-01-22 09:09:37
中国中医药现代远程教育(2014年11期)2014-08-08 13:23:44
疑难病杂志(2014年12期)2014-04-16 05:19:33
中国中医药现代远程教育(2014年17期)2014-03-01 04:29:20
