
高糖环境下法舒地尔调控自噬对原代心肌细胞凋亡的作用
2023-06-28 07:53高惠宽
医学研究杂志 2023年6期
高惠宽 侯 斐
糖尿病心肌病是一种主要的糖尿病并发症,是糖尿病患者的主要死因之一。心肌细胞凋亡、心肌纤维化是糖尿病心肌病的主要病理状态,可导致心脏舒张和收缩功能障碍。抑制心肌细胞凋亡的药物可以预防糖尿病心肌病。
细胞凋亡在很大程度上受Bcl家族基因的调控,包括抗凋亡蛋白Bcl-2和促凋亡蛋白Bax。既往研究表明,无论是在体内还是体外,抑制Rho激酶(Rho kinase,ROCK)活性或下调ROCK都能抑制病理刺激诱导的心肌细胞凋亡[1,2]。法舒地尔是一种ROCK抑制剂,有研究表明,其可以改善糖尿病患者蛋白尿情况,亦可以抑制过度耐力运动训练引起的心肌细胞凋亡[3,4]。但法舒地尔对糖尿病心肌细胞凋亡的影响及其机制尚不清楚。自噬是细胞能量代谢和细胞存活最重要的调节靶点之一。有研究发现,糖尿病肾病小鼠模型中,肾组织中自噬蛋白表达增多[5]。然而,自噬在促进或改善糖尿病心肌病细胞凋亡中的作用仍未得到阐明。一些研究表明,法舒地尔可增强自噬[6,7]。本研究旨在探讨高糖环境下,法舒地尔对原代心肌细胞凋亡的作用及自噬在其中的作用。
材料与方法
1.主要试剂:DMEM培养基、胎牛血清FBS购自美国Gibco公司;盐酸法舒地尔注射液购自天津红日药业股份有限公司;3-甲基腺嘌呤(3-methyladenine,3-MA)、兔抗鼠P62抗体、兔抗鼠α-横纹肌肌动蛋白(α-SA)抗体、Trizol购自美国Sigma公司;兔抗鼠肌球蛋白磷酸酶靶标亚基1(MYPT1)抗体、兔抗鼠磷酸化(p)-MYPT1抗体、兔抗鼠Bcl-2抗体、兔抗鼠Bax抗体、兔抗鼠LC3抗体购自英国Abcam公司;异硫氰酸荧光素(fluorescein isothiocyanate,FITC)-膜联蛋白V(Annexin V)检测试剂盒购自美国Thermo Fisher Scientific公司。
2.乳鼠心肌细胞原代分离与培养:实验方案经首都医科大学附属北京友谊医院医学伦理中心批准(伦理学审批号:16-5001)。出生2天乳鼠购自北京维通利华实验动物技术有限公司。腹腔注射10%水合氯醛,按体质量计算用量,0.1ml/10g。无菌条件下开胸取出C57BL/6乳鼠心脏,去除心房,用PBS多次洗净血液,之后剪碎,加入0.2%的Ⅱ型胶原酶,37℃水浴震荡40~60min,之后吹打离心1200r/min,后DMEM完全培养基重悬种,采用差速贴壁获得心肌细胞(乳鼠差速2h)。上清液吸出(内含心肌细胞),上清液用Hanks液清洗、离心,共 3次,获得心肌细胞[8]。原代培养心肌细胞,第2~3代细胞用于实验。
3.原代心肌细胞HE染色:心肌细胞接种于96孔细胞培养板中;培养2~3天后,PBS清洗;4%多聚甲醛固定20min;苏木精染色2~5min,水洗2min;盐酸分化至细胞核染色清晰,水洗,并浸于水中至核变蓝;伊红液染色2min,水洗2min;脱水透明;中性树胶封片。
4.原代心肌细胞免疫鉴定:心肌细胞接种于96孔细胞培养板中;培养2~3天后,PBS清洗;4%多聚甲醛固定20min,PBS清洗;0.1% TritonX-100室温孵育30min,PBS清洗;H2O2室温孵育10min,PBS清洗;1%FBS室温封闭1h;滴加α-SA抗体,4℃过夜;PBS清洗,滴加荧光标记的二抗,室温避光孵育1h;PBS清洗,DAPI染核室温避光孵育15min,PBS洗3次;荧光显微镜下观察荧光。
5.细胞培养及分组:细胞培养于含10%胎牛血清、1%双抗(青霉素、链霉素)的低糖(葡萄糖5.5mmol/L)DMEM培养基中,当细胞达到80%~90%融合时传代或给药处理。给药前,将细胞接种于6孔板(每孔约105个细胞),于含10%胎牛血清的低糖DMEM培养基中培养24h;后更换培养基为含1%胎牛血清的低糖DMEM培养基,继续培养12h。此后,按照以下分组更换培养基及加入药物:对照组(LG组,培养基葡萄糖浓度5.5mmol/L)、高糖组(HG组,葡萄糖浓度25mmol/L)、LG+法舒地尔组(LF组,加入100μmol/L法舒地尔)、HG+法舒地尔组(HF组)、高糖+法舒地尔+3-MA组(HM组,加入5mmol/L 3-MA)。给药处理后,各组细胞继续培养24h。
6.流式细胞术实验:把细胞培养液吸出至离心管内。冰PBS洗涤贴壁细胞1次,加入适量胰酶消化细胞。消化好后,加入之前收集的细胞培养液,轻轻吹打均匀,转移到离心管内,1000×g离心5min;PBS洗涤细胞两次并计数,收集(1~5)×105细胞,然后1000×g离心5min;加入100μl×Binding Buffer重悬细胞;加入5μl AnnexinV-FITC、5μl PI染色液,室温避光孵育15min;加入400μl Binding Buffer;上流式细胞仪检测。
7.Western blot法检测蛋白表达:各组细胞弃去培养上清,用PBS洗两次,再加入150μl裂解液。冰上裂解40min,4℃下12000r/min离心15min。BCA法蛋白定量,电泳转膜至NC膜。奶粉封闭的膜用5%BSA-TBST稀释一抗,4℃过夜,滴加二抗,山羊抗兔IgG(H+L)HRP 1∶1000,山羊抗鼠IgG(H+L)HRP 1∶1000室温孵育40min。TBST洗膜后,ECL滴加到膜的蛋白面,反应2min,做化学发光。将膜放到化学发光仪中收集信号。
8.RT-PCR实验:各组细胞弃去培养上清,加入Trizol变性液,提取细胞中总RNA,待RNA完全溶解后检测其浓度。采用两步法,以β-actin为内参体系,进行实时荧光定量PCR。反应程序:95℃ 3min,95℃ 30s,55℃ 20s,72℃ 20s共40个循环;溶解曲线步骤:95℃ 15s,60℃ 15s,20min升温,95℃ 15s。目的基因引物序列,ROCK1上游引物:5′-CAAGTCAGACCTCACAGCTT-3′,下游引物:5′-CTCATCTCTGTGTGACTCTT-3′;ROCK2上游引物:5′-AGCTGGAAAGAGAAAAGGCC-3′,下游引物:5′-TGTTTCTGGAGCTGATTGAC-3′。统计学分析采用相对定量,应用RQ=2-△△CT进行计算相对表达量。
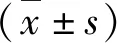
结 果
1.原代心肌形态及免疫鉴定:原代心肌呈圆形或梭形(图1A),α-SA抗原表达呈阳性反应(图1B)。
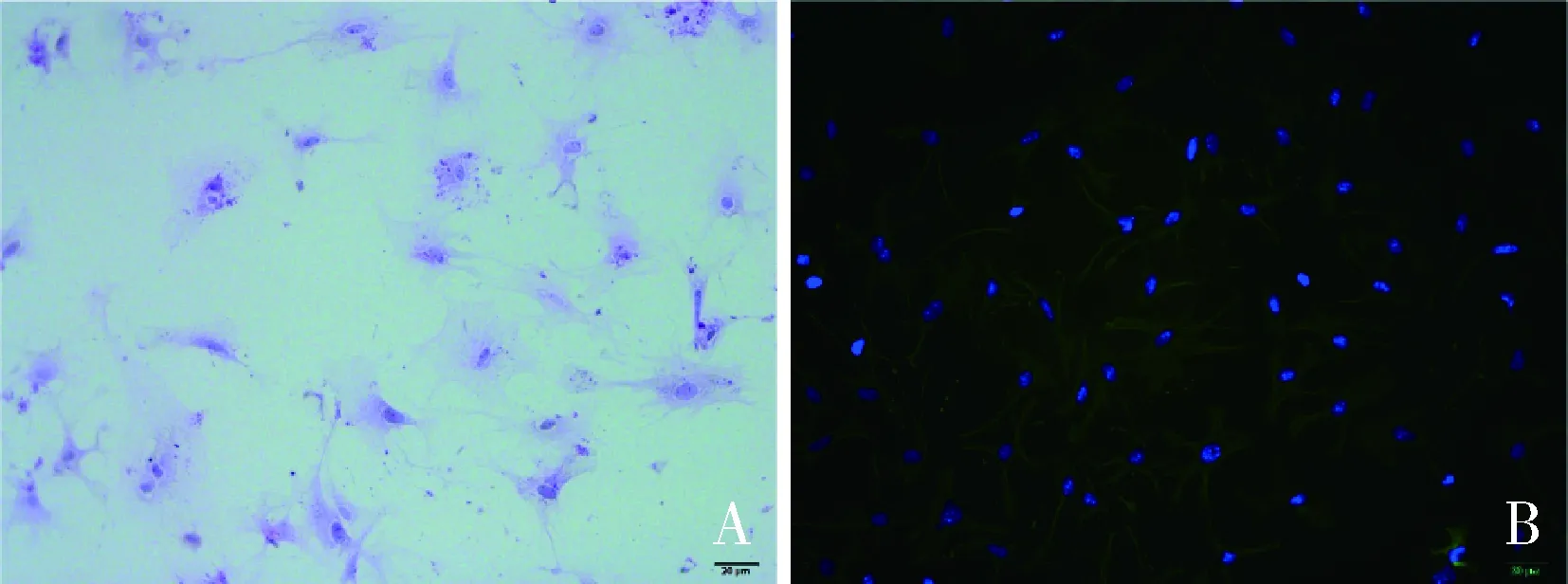
图1 原代心肌细胞形态及免疫鉴定(×100)A.HE染色;B.α-SA免疫荧光染色(绿色为α-SA阳性染色,蓝色为细胞核)
2.法舒地尔抑制高糖环境下的原代心肌细胞凋亡:使用Annexin V-FITC、PI标记细胞,采用流式细胞术检测早期凋亡细胞比例。高糖组细胞凋亡比例较低糖组明显升高(53.4% vs 1.0%)。使用法舒地尔干预后,细胞凋亡比例下降至13.9%。而3-MA干预可逆转法舒地尔引起的细胞凋亡的下降(13.9%升至37.2%,图2)。笔者检测了各组细胞抗凋亡蛋白Bcl-2及促凋亡蛋白Bax的表达情况,结果显示,高糖组Bcl-2表达降低、Bax表达升高,而法舒地尔干预后可部分逆转该变化(HF组 vs HG组),而3-MA干预可抑制法舒地尔的作用(HM组 vs HF组,图3)。
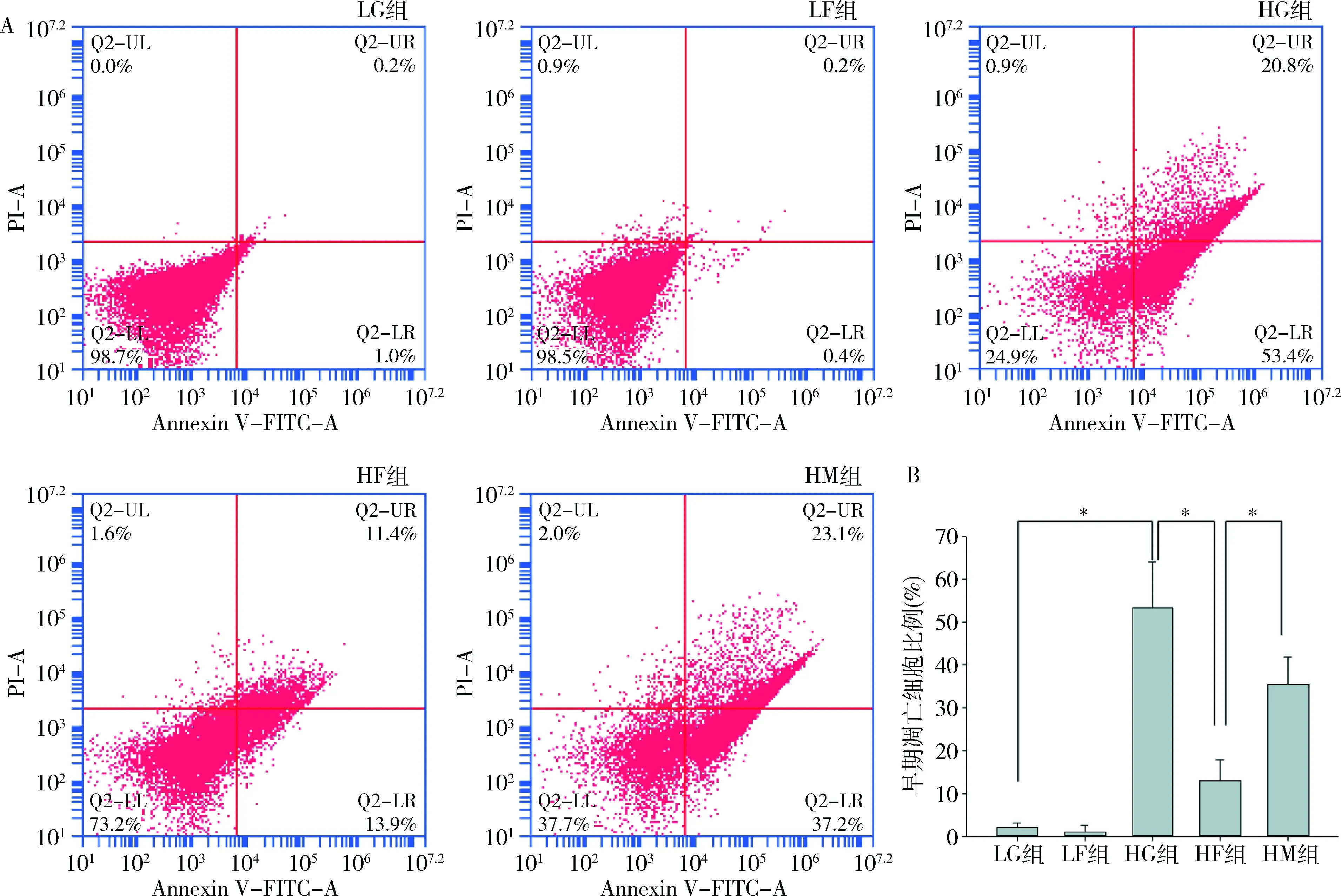
图2 法舒地尔抑制高糖环境下原代心肌细胞凋亡,3-MA可逆转法舒地尔的作用A.流式细胞术检测细胞凋亡情况:Q2-UL坏死细胞,Q2-UR晚期凋亡细胞,Q2-LL活细胞,Q2-LR早期凋亡细胞;B.各组早期凋亡细胞比例,计算Q2-LR区细胞数与4区所有细胞数的比值即为早期凋亡细胞比例,*P<0.05
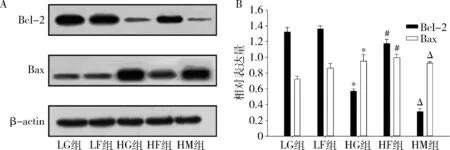
图3 法舒地尔及3-MA对高糖环境下原代心肌细胞凋亡相关蛋白Bcl-2、Bax的影响A.Western blot法检测结果;B.各组细胞Bcl-2、Bax的相对表达量。与LG组比较,*P<0.05,**P<0.01;与HG组比较,#P<0.05;与HF组比较,△P<0.05
3.法舒地尔抑制高糖环境下原代心肌细胞内ROCK表达:测定ROCK1、ROCK2的mRNA表达反映ROCK表达情况,同时采用Western blot法检测ROCK下游蛋白MYPT1及p-MYPT1的表达量,计算p-MYPT1/MYPT1比值反映ROCK活性变化。高糖环境下ROCK1、ROCK2 mRNA表达增加、p-MYPT1/MYPT1比值升高(HG组 vs LG组),而法舒地尔可使ROCK1、ROCK2 mRNA表达减少、p-MYPT1/MYPT1比值降低(HF组 vs HG组,HM组 vs HG组)。这些结果表明,法舒地尔抑制高糖环境下原代心肌细胞内ROCK表达(图4)。
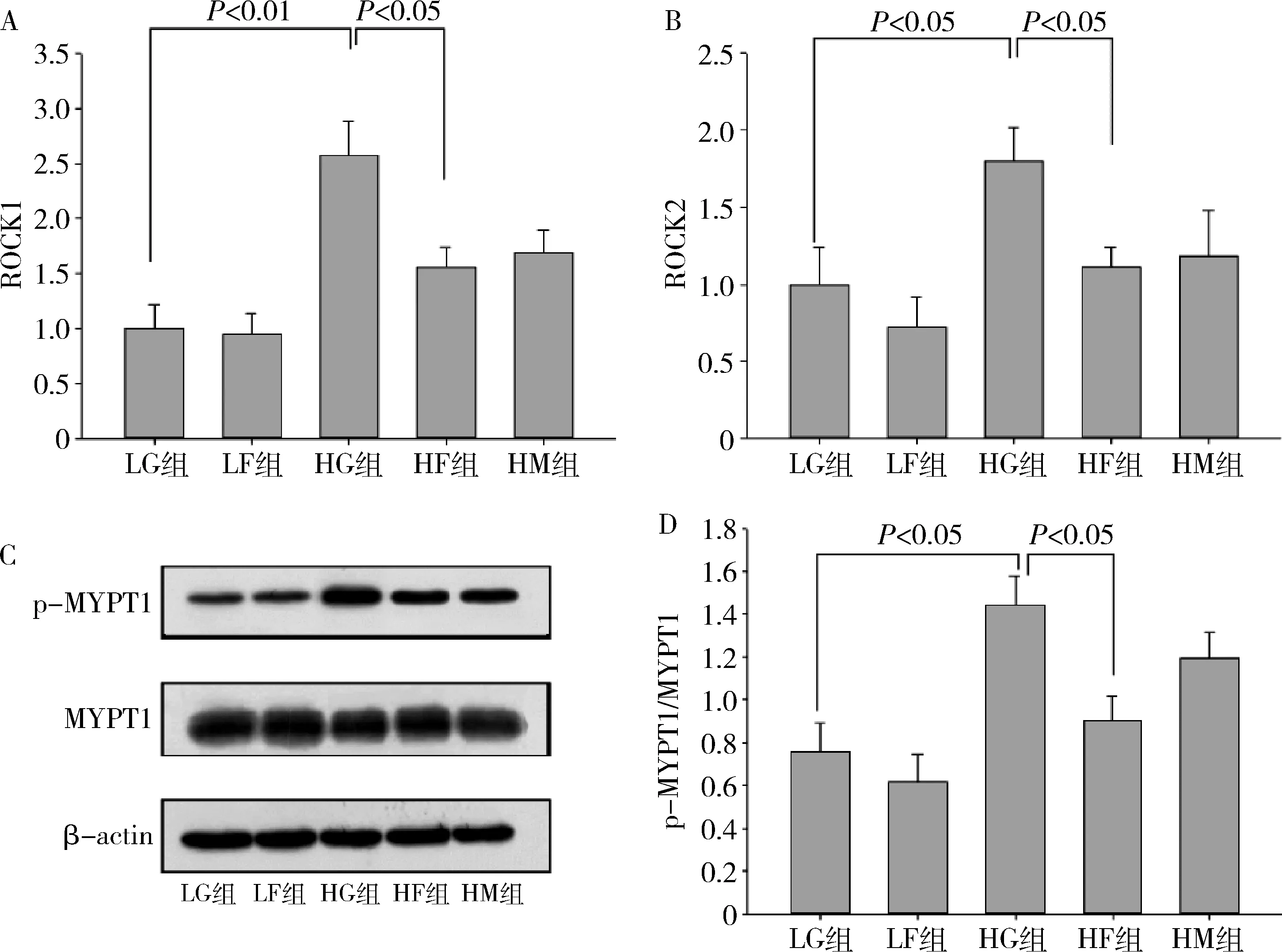
图4 法舒地尔及3-MA对高糖环境下原代心肌细胞ROCK信号通路的影响A.ROCK1 mRNA相对表达量(RT-PCR法);B.ROCK2 mRNA相对表达量(RT-PCR法);C.Western blot法检测p-MYPT1、MYPT1蛋白情况;D.p-MYPT1/MYPT1蛋白相对表达量比值
4.法舒地尔活化了高糖环境下原代心肌细胞内受阻的自噬流:高糖组心肌细胞内不可溶性P62蛋白表达升高,提示自噬流受阻。使用法舒地尔干预后,不可溶性P62蛋白表达减少、可溶性P62蛋白表达增多、同时LC3-Ⅱ/LC3-Ⅰ增大,说明自噬流活化。而使用自噬抑制剂3-MA,可使自噬流阻滞,逆转法舒地尔的作用(图5)。
讨 论
心肌细胞凋亡后继发心脏成纤维细胞活化导致心脏纤维化是糖尿病心肌病左心室舒张和收缩功能障碍的主要原因。与正常人比较,糖尿病患者心肌细胞、内皮细胞及心脏成纤维细胞的凋亡数分别为正常人的85、61及26倍[9]。这些结果说明心肌细胞对糖尿病诱导的细胞凋亡最敏感。本研究采用流式细胞术检测细胞凋亡率,高糖组细胞凋亡率较低糖组明显升高,所以高糖可以引起心肌细胞凋亡。
Rho GTP酶通过激活其下游靶分子ROCK调节细胞的多种行为与功能。ROCK有两种亚型,即ROCK1和ROCK2。近年来分析发现,心脏中ROCK2表达为主。法舒地尔是ROCK的非亚基选择性的抑制剂,既抑制ROCK1也抑制ROCK2的表达。ROCK的作用底物有很多,典型的是MYPT1。本研究可以见到高糖可以激活RhoA/ROCK信号转导通路,法舒地尔可抑制RhoA/ROCK的表达。研究发现,阻断RhoA/ROCK信号转导通路可减轻细胞凋亡,可能通过调控Bcl-2/Bax、caspase-3、caspase-2的表达[2,10~13]。本研究在高糖培养的原代心肌细胞中使用法舒地尔干预后,细胞凋亡可减轻。说明法舒地尔可以抑制高糖环境下的心肌细胞凋亡,而且,法舒地尔对细胞凋亡的作用机制既有促凋亡蛋白Bax的减少,亦有抗凋亡蛋白Bcl-2的增加。
细胞凋亡和自噬作为调控细胞程序性死亡的两种重要方式,广泛存在于真核细胞的生理病理过程中。尽管凋亡与自噬的特征及机制不同,但两通路并不是独立并行的,两者有共同的刺激因子和调节蛋白,两通路之间存在着错综复杂的对话。众多调控元件参与凋亡和自噬的交互应答机制,包括 Beclin1、Bcl-2家族蛋白、P53等。有研究发现,增强自噬可减轻细胞凋亡水平[14]。国内有研究发现,二甲双胍可通过增强细胞自噬减轻糖尿病大鼠血管内皮细胞凋亡[15]。低糖或低氧环境下,心肌细胞自噬活性增强、细胞凋亡比例减少[16~18]。目前,国内外关于Rho/ROCK信号通路与自噬信号通路直接关系较少。Gurkar发现ROCK1可以磷酸化Beclin1的Thr119,引起Beclin1/Bcl-2复合体分离,从而引起自噬增强。然而亦有研究表明法舒地尔阻断ROCK,可增强自噬[6,7]。
P62是自噬过程中最重要的转运蛋白,它是连接LC3与待降解产物的中介。P62蛋白含量变化反应自噬流的活化与否,自噬流活化时,P62蛋白含量降低;自噬流抑制时,P62蛋白含量增加。实际操作中,可同时观察可溶性P62蛋白、不可溶性P62蛋白和LC3-Ⅱ/LC3-Ⅰ转化综合判断自噬流状态。若可溶性P62蛋白减少,不可溶性P62蛋白无明显改变,同时LC3-Ⅰ向LC3-Ⅱ转化增加则表明自噬流活化;若可溶性P62蛋白减少,但不可溶性P62明显增加,则无论LC3的变化,均表明自噬流阻断。本研究发现高糖引起心肌细胞内不可溶性P62增加,提示自噬流阻断;使用法舒地尔干预后,可溶性P62蛋白减少同时伴有LC3-Ⅱ/LC3-Ⅰ升高,说明自噬流活化。而使用自噬抑制剂3-MA阻断自噬流后(不可溶性P62蛋白明显升高),法舒地尔抑制心肌细胞凋亡的作用被部分反转,说明自噬流的活化在法舒地尔对高糖环境下心肌细胞凋亡中起着重要的作用。
综上所述,本研究以离体培养的原代心肌细胞为研究对象,使用高糖培养基,并使用法舒地尔及自噬抑制剂干预,表明法舒地尔通过活化自噬流抑制高糖环境下心肌细胞凋亡。今后将继续探讨自噬在糖尿病心肌病中的分子生物学机制,并进行动物实验进一步加以验证。该研究拟为糖尿病心肌病的治疗提供新的理论依据和实验数据。
猜你喜欢
解放军医学院学报(2020年12期)2020-03-29
心肺血管病杂志(2019年9期)2019-12-09
中成药(2018年9期)2018-10-09
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
中国继续医学教育(2015年5期)2016-01-07
中国药理学与毒理学杂志(2015年3期)2015-12-16
医学研究杂志(2015年9期)2015-07-01
长江蔬菜(2015年3期)2015-03-11
中华皮肤科杂志(2014年3期)2014-12-19
