
基于机器学习的成纤维细胞生长因子受体激酶抑制剂虚拟筛选模型
2023-06-25 09:56丁俊涛刘博吴建章李物兰
温州医科大学学报 2023年7期
丁俊涛,刘博,吴建章,李物兰
1.温州医科大学 第一临床医学院(信息与工程学院),浙江 温州 325035;2.温州医科大学附属眼视光医院,浙江 温州 325000
成纤维细胞生长因子受体(fibroblast growth factor receptors, FGFR)是一种在许多生物学过程中发挥重要作用的受体酪氨酸激酶,包括FGFR1、FGFR2、FGFR3、FGFR4四种亚型[1]。FGFR信号传导的异常激活在不同类型肿瘤(如胆管癌、子宫内膜癌、尿路上皮癌和肺癌等)的发生和进展中起重要作用[2-4],FGFR抑制剂具有治疗这些疾病的潜力。目前FGFR激酶家族中已有3种小分子靶向抑制剂在近三年被批准用于癌症治疗[5],但是随后发现它们均表现出较强的高血磷症、腹泻等不良反应,因此急需寻找出新颖先导化合物骨架用于改构成安全性更好的抑制剂。
在新药发现领域,与实验筛选相比,虚拟筛选方法在经济成本、时间效率上极具优势[6]。目前虚拟筛选可分为传统经典的计算机辅助药物设计(computer aided drug design, CADD)[7]和现代新颖的人工智能药物设计(artificial intelligence drug design, AIDD)[8]。在面对千万级以上大数据库的虚拟筛选方面,AIDD的效率远高于CADD。在已报道的FGFR抑制剂中,部分是基于CADD的新药发现,未见基于AIDD的抑制剂研究。因此,本研究建立了基于AIDD的FGFR激酶抑制剂虚拟筛选模型,选择FGFR四种受体中与肿瘤等疾病关系密切的FGFR1,将AIDD筛选得到的化合物用CADD进一步进行了FGFR1激酶抑制剂的虚拟筛选和分子动力学模拟研究,旨在为FGFR抑制剂的研究提供高效AIDD筛选模型和苗头化合物。
1 材料和方法
1.1 FGFR激酶抑制剂数据整理由于FGFR1-4激酶结构,尤其是FGFR1-3,高度相似,因此目前已报道的FGFR激酶抑制剂绝大部分是泛FGFR抑制剂。将BindingDB公共药物实验数据集[9]导入Mysql[10]软件进行整理查询,获得2196条FGFR激酶抑制剂实验数据,并将半数抑制浓度小于100 nmol/L的记为活性数据,标签值为1,其余的记为非活性数据,标签值为0。在对数据进行清洗、去重、标签等操作后,最终得到活性数据1275条,非活性数据921条,作为后续处理和训练的数据集合。
1.2 分子的特征工程采用两种分子特征表示方法:①分子指纹MACCS[11]:包含166位分子指纹,指纹中的每一位都代表特定的子结构,可使用RDKit 根据分子的简化分子线性输入规范(simplified molecular input line entry system, SMILES)[12]计算获得;②分子描述符:使用RDKit根据分子SMILES计算获得,内容包括分子量、脂水分配系数、拓扑极性表面积和可旋转键数等,从不同角度描述分子性质,RDKit描述符合共有206维。
1.3 基于机器学习虚拟筛选模型的构建采用随机森林和支持向量机两种机器学习方法建立虚拟筛选模型:①随机森林:建立由决策树组成的“森林”,利用多棵决策树的结果对样本进行训练并预测的分类器,通过Sklearn[13]完成模型建立并对随机森林超参数进行调试和训练。②支持向量机:使用监督学习模式对输入数据进行二元分类的线性分类器,其算法核心思想是求解输入数据的最大边距超平面以完成分类。使用Sklearn完成模型构建并完成训练测试。随机森林和支持向量机两种模型的构建都通过Python和Sklearn完成,模型的整体结构包括化合物特征提取、数据载入、划分测试集和验证集、模型拟合训练、参数迭代调整以及最终的活性预测。
1.4 基于机器学习虚拟筛选模型的评价指标使用准确率、精准率、召回率、曲线下面积(area under curve, AUC)4个指标验证机器学习模型在数据集上的综合性能。对于二分类问题,预测结果有4种划分,分别是:真阳(true positive, TP)、真阴(true negative, TN)、假阳(false positive,FP)、假阴(false negative, FN)。本研究中所用的指标计算方法如下:
准确率:所有预测正确(TP与TN)占总的比率,用于判断模型预测分类是否准确。
精准率:预测为正且实际为正占全部预测为正的比率。
召回率:预测为正且实际为正占全部实际为正的比率。
AUC:对受试者工作特征曲线(receiver operating characteristic curve, ROC)进行积分计算,用来定量描述分类器的好坏,AUC的值越大则分类性能越强。
1.5 基于分子对接的虚拟筛选在用机器学习模型筛选获得潜在的活性化合物后,选择FGFR家族中的FGFR1,进一步采用两级递进的基于分子对接的虚拟筛选,即依次用Autodock Vina软件[14]和Glide 方法的XP(Extra Precision)精度进行FGFR1激酶抑制剂的虚拟筛选。使用的模板蛋白结构从PDB网站获取,蛋白结构的PDBID为4V05[15]。对蛋白结构模型进行加氢、能量优化等操作后,以模型内包含的AZD4547的结合位置为中心,选取60 Å×60 Å× 60 Å大小的格子作为对接的区域进行虚拟筛选。
1.6 分子动力学模拟使用Gromacs2020.4软件进行分子动力学模拟,使用Charmm36力场[16]为复合体系的每个蛋白质生成拓扑和参数文件,小分子配体使用CGenff力场参数。然后将体系溶解于TIP3P十二面体水盒中,距离体系表面10 Å处,加入适量的反离子使体系中和,接着采用最陡下降法进行能量最小化50000步,然后进行100 ps的NVT (Canonical Ensemble,恒定粒子数、体积和温度)模拟和100 ps的NPT(Constant-pressure Constant-temperature,恒定粒子数、压力和温度)模拟,温度限制在300 k,最后用MD模拟每个系统的NPT,持续时间为500 ns。所有仿真步骤均为 2 fs,轨迹每2 ps保存1次,以供后续分析。
2 结果
2.1 BindingDB中活性化合物理化性质分析对BindingDB库内实验数据进行分析,库内化合物分子质量的分布有一定的差异(见图1A),活性化合物的分子量分布大多集中在442~557之间,且其中位数略高于非活性化合物,符合类药性五原则对于分子量的要求。化合物的拓扑极性表面积 (topological polar surface area, tPSA)分布也有一定差异(见图1B),活性化合物的tPSA较高,集中在89~117之间。另外,活性化合物的定量评估类药性(quantitative estimate of drug-likeness,QED)多集中于0.5,而非活性化合物集中于0.4(见图1C),说明活性化合物有更好的成药性。最后,活性化合物结构中成环多数为5个,而非活性化合物则为4个(见图1D)。环的数量也可以导致化合物亲疏水性质和与靶点亲和力的变化。

图1 BindingDB数据库FGFR1实验数据理化性质统计分析
2.2 模型评价通常认为,准确率和AUC值大于0.75,分类器有较好的准确性和分类效果。在FGFR激酶抑制剂的模型中,随机森林相比于支持向量机在准确率、精准率、召回率有更好的表现(见表1)。从ROC曲线来看,随机森林拐点更靠近左上角(见图2),说明随机森林算法有更好的预测性能。
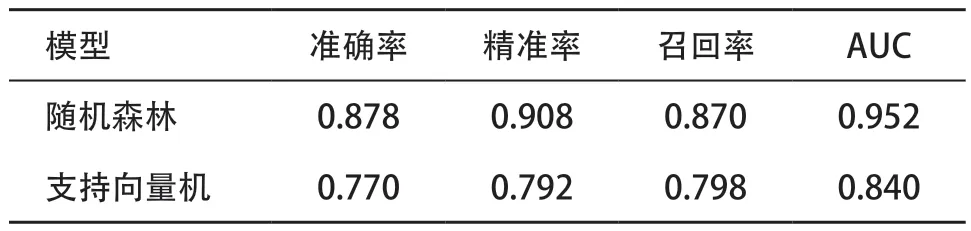
表1 机器学习模型评价指标

图2 机器学习模型的ROC曲线
2.3 虚拟筛选结果虚拟筛选的化合物库来自陶素公司提供的包含1300万个小分子的虚拟化合物库。针对该库使用随机森林方法进行第一级的虚拟筛选,将预测阳性概率为0.999以上的化合物认为是潜在的活性化合物,共计筛选获得10340个潜在的活性化合物,约占总数的0.7%,耗时约28.3 h。 随后对10340 个化合物用Autodock Vina软件和Glide方法逐级筛选FGFR1激酶抑制剂。用Autodock Vina软件得到打分值小于-9.00 kcal/mol的分子共395个,占比3.8%。用Glide法从395个化合物中筛选得到3个打分小于-9.80 kcal/mol的化合物,占比0.76%。其对接打分值如表2所示,最优化合物的打分结果为-11.12 kcal/mol。AZD4547(见图3A)、化合物a(见图3B)、化合物b(见图3C)均与FGFR1激酶ALA-101形成氢键作用,分子都伸入口袋内部疏水口袋,化合物a、化合物b以及AZD4547与FGFR1结合模式相同[17]。化合物a和b与阳性对照AZD4547一样与FGFR1激酶蛋白有着较多的氢键相互作用,不同的是,化合物c仅由疏水作用维持结合(见图3D),没有与FGFR1形成氢键相互作用,也没有卤键等相互作用,导致其与FGFR1的结合效果较差。此 外,化合物a上Cl和F分别和残基LYS-19、VAL-29形成卤键,推测与FGFR1的结合力较AZD4547更好。
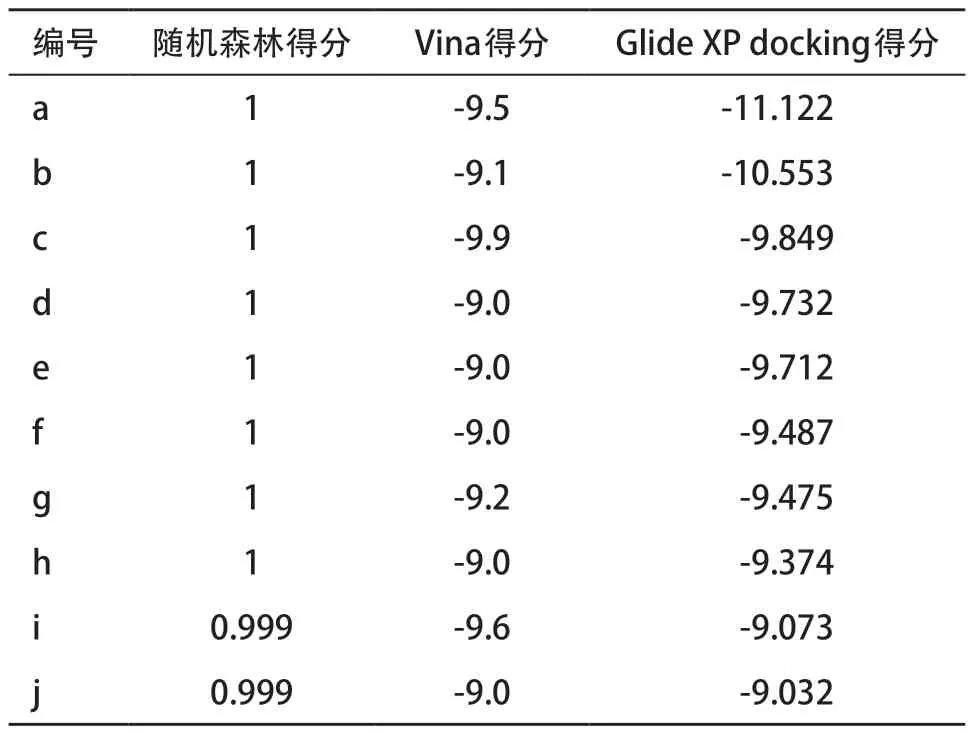
表2 随机森林、Vina和Glide XP虚拟筛选结果
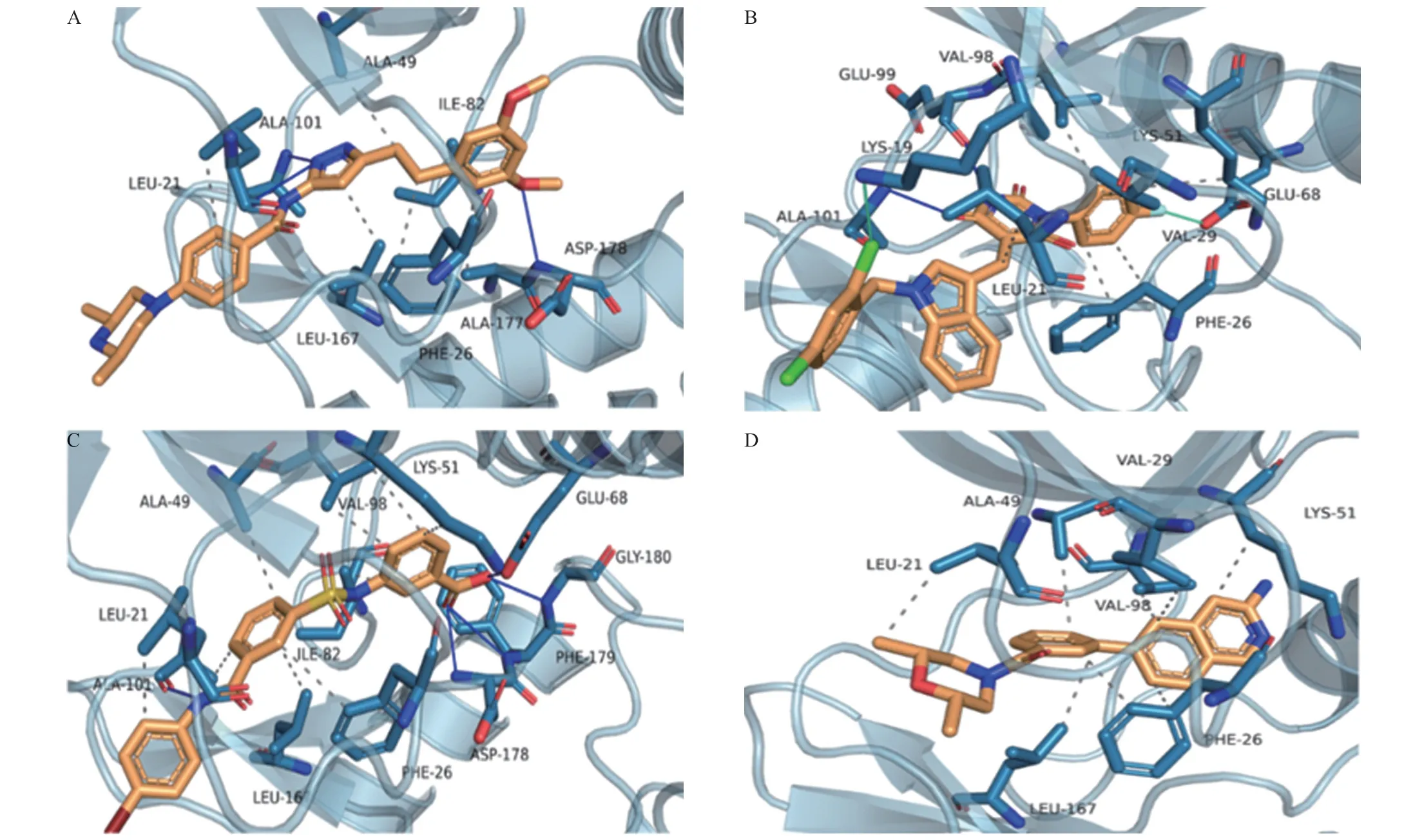
图3 FGFR1激酶(PDBID为4V05)与AZD4547(A)、化合物a(B)、化合物b(C)、化合物c(D)的结合模式
2.4 分子动力学模拟结果将模板蛋白的配体AZD4547 与打分最高的3 个潜在活性化合物进行 100 ns的分子动力学模拟分析。通过MM/GBSA方法对4个体系的较为稳定的时间段70~80 ns进行结合自由能计算(见图4、表3),其中AZD4547结合自由能最优,总的结合自由能为-46.72 kcal/mol,化合物a总的结合自由能为-38.13 kcal/mol,化合物b总的结合自由能为-42.87 kcal/mol,化合物c总的结合自由能为-13.68 kcal/mol,结合图3中4个化合物的结合模式,说明氢键和卤键是提升化合物与FGFR1亲和力的关键,化合物d与FGFR1没有氢键作用,所以结合自由能较高。对比结合口袋位置氨基酸残基能量贡献情况(见图5),其中LEU21、VAL29、ALA49这3个残基在4个体系中都贡献了较多的结合自由能,从结合自由能角度解释了化合物a、b、c的结合情况与AZD4547较为相似。但是化合物c与其他化合物不同的是,ASP178削弱了结合自由能,这也是化合物c结合自由能较差的一个关键因素。对比4组体系的均方根偏差(root mean square deviation,RMSD)(见图6),4个小分子化合物在口袋内较为稳定,其中化合物a较AZD4547有更好的稳定性,其RMSD波动范围较小;化合物b与AZD4547相比波动性较大,但波动范围均未超过0.4 Å;化合物c稳定性最差,它们的RMSD结果与前期分子对接的结果相吻合。
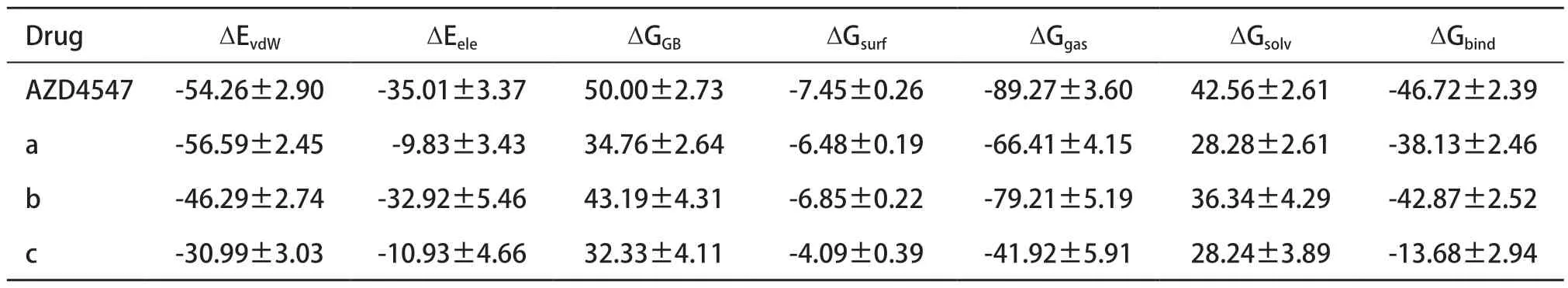
表3 FGFR1与4种化合物结合的自由能情况(kcal/mol)
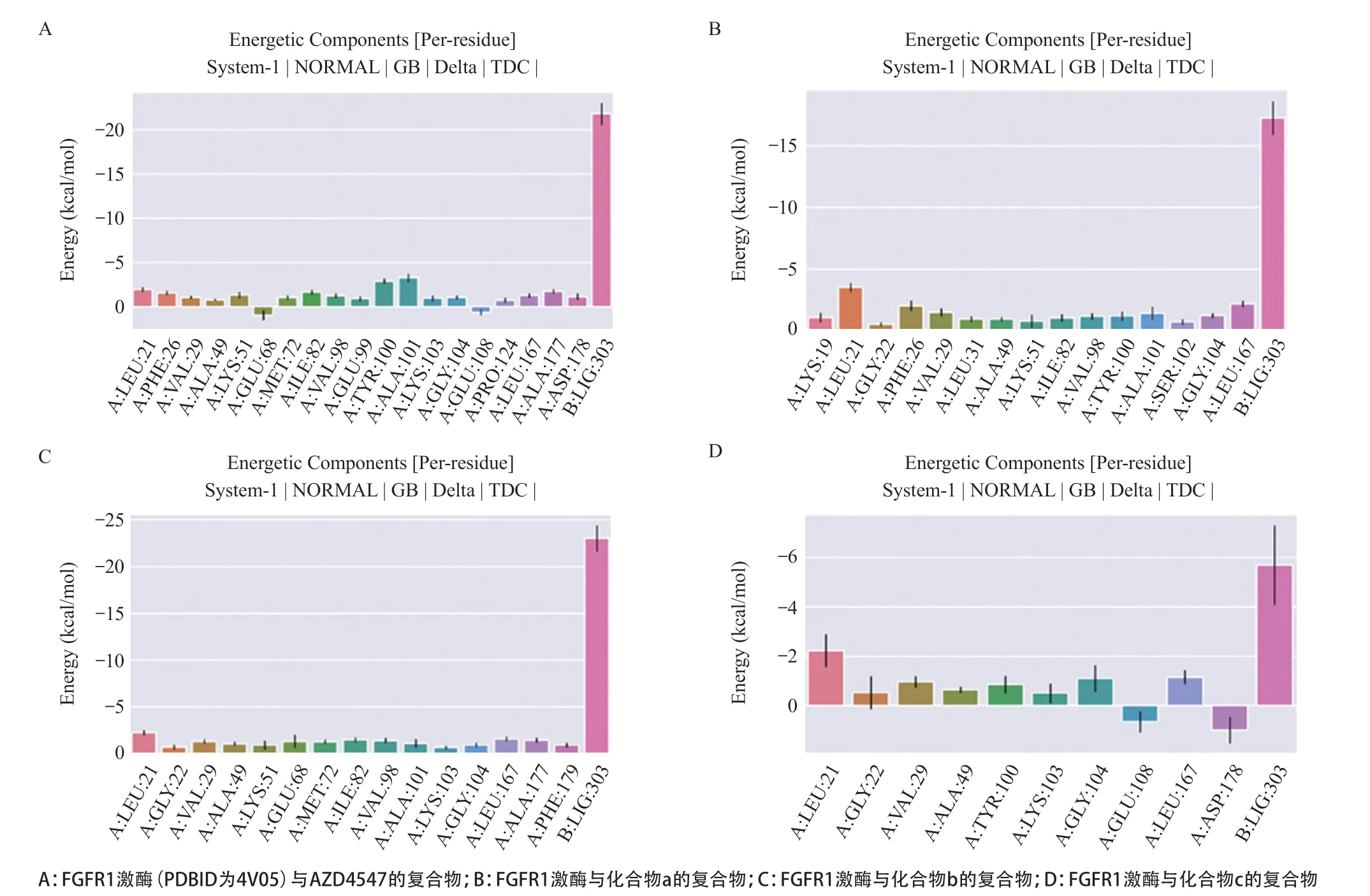
图5 FGFR1激酶化合物复合体系在70~80 ns时氨基酸贡献自由能统计

图6 FGFR1激酶化合物体系分子动力学模拟的RMSD
3 讨论
近年由于人工智能的蓬勃发展,越来越多的机器学习和深度学习技术被应用于药物发现。在激酶领域中,受体酪氨酸激酶家族已有较为成功的人工智能结合发现药物案例。在人工智能与新药发现 中,主要困难在于药物活性数据与阴性数据的缺乏,目前较为成功的案例如血管内皮生长因子受体、表皮生长因子受体往往是具备较多实验测试数据的靶点[18-20]。而本研究的FGFR抑制剂目前数据相对缺少,现有数据库中活性数值模糊,对该领域机器学习发展有较大掣制,且尚未见有利用人工智能发现FGFR抑制剂的相关报道。因此本研究在数据准备时,对已有药物数据库和文献抑制剂数据进行整理,并将其用于人工智能模型构建和药物筛选。本研究中使用的随机森林和支持向量机两种机器学习模型的准确率分别为0.878和0.770,AUC分别是0.952和0.840,这表明本研究构建的模型较好。与支持向量机算法构建的模型相比,随机森林构建的模型在准确率、精准率、召回率方面总体更优。同时机器学习模型从1300万个小分子化合物库筛选获得10340个潜在的活性化合物,耗时约28.3 h,这说明机器学习模型在面对较大虚拟筛选药物库时可以快速准确地从中筛选出可能的先导化合物,快速缩小筛选范围,节约时间和经济成本,提高效率。
分子动力学模拟是一种有效分析小分子化合物与激酶结合模式和结合能力的方法。通过分子动力学模拟并计算激酶和化合物的结合自由能,a和b的图6 FGFR1激酶化合物体系分子动力学模拟的RMSD结合自由能与AZD4547相近,在理论上说明化合物a、b可能有接近AZD4547的抑制活性。将结合自由能分解,最优的3个化合物的结合自由能主要贡献来源于范德瓦尔斯力,其次是静电力,由于化合物a的静电力贡献较小,导致其最终的自由能情况较差。这说明对化合物的改造可以通过增强静电力的贡献和维持疏水作用来提升化合物抑制活性。对化合物周围的氨基酸残基能量贡献进行分析,在整个过程中,LEU21、ALA101两个残基对结合能的贡献在几个化合物体系中均有呈现,说明LEU21、ALA101两个残基是主要影响该系列先导化合物和FGFR1结合的关键残基,在化合物b和FGFR1的复合体系中,GLU108和ASP178两个残基对结合能的贡献起到反作用,这说明这两个残基可能阻碍化合物和FGFR1结合,因此化合物b的结合能较其他3个化合物相比更高。上述结果表示该模型的虚拟筛选性能较好。
总之,本研究提供了一种基于机器学习的FGFR激酶抑制剂虚拟筛选模型,其可以短时间内高效筛选规模较大的小分子库,快速高效地获得潜在活性的FGFR激酶抑制剂。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
上海农业学报(2017年3期)2017-04-10
池州学院学报(2015年3期)2016-01-05
中国医药生物技术(2015年4期)2015-12-26
天津科技大学学报(2015年2期)2015-08-09
中国当代医药(2015年16期)2015-03-01
中国药理学通报(2014年2期)2014-05-09
